




Modern tools of biological investigation give us opportunities to develop drugs much more efficiently. The president of Europes most important trials organisation explains how these opportunities can and must be exploited to start delivering drugs that are more effective and more affordable.
 The road to developing a new medicine – translating a new idea into a drug licensed to treat patients – is long, often too long. Traditional drug development moves through preclinical studies to phase I, II and III trials, with increasing resources needed for each stage, from 25–30% of costs for preclinical work to the bulk of 40% for phase III late-stage development. The attrition rate is enormous. For every 10,000 compounds screened, only one will make it successfully to the clinic. But are we really sure that the remaining 9,999 others are really not useful in any way? The current approach means we don’t know how many potentially useful compounds we may have missed.
The road to developing a new medicine – translating a new idea into a drug licensed to treat patients – is long, often too long. Traditional drug development moves through preclinical studies to phase I, II and III trials, with increasing resources needed for each stage, from 25–30% of costs for preclinical work to the bulk of 40% for phase III late-stage development. The attrition rate is enormous. For every 10,000 compounds screened, only one will make it successfully to the clinic. But are we really sure that the remaining 9,999 others are really not useful in any way? The current approach means we don’t know how many potentially useful compounds we may have missed.
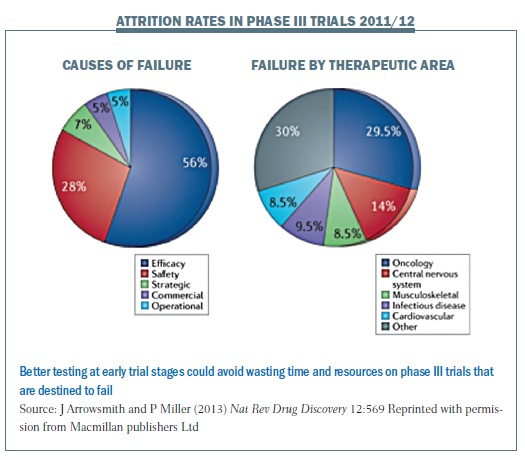
The figure below shows the attrition rates for therapies in recent phase III trials, with nearly 60% failing due to lack of efficacy (Nat Rev Drug Discovery 2013; 12:569). Oncology is the leading therapeutic area for late-stage failures. We’re doing something wrong when we fail so late, and particularly in oncology if we fail more often than in other areas.
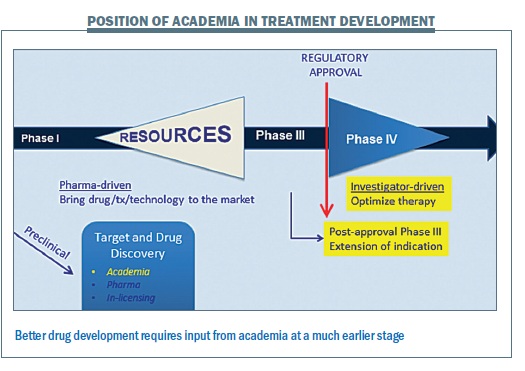
 Academia is currently involved rather late in the traditional drug development model (see below), usually at phase III for larger trials or at phase IV for investigator-driven optimisation or extension of indication after approval, with perhaps some work in target and drug discovery. Everything in between is largely led and organised by pharmaceutical companies, but I think there are opportunities for academics to contribute more in the earlier stages of clinical development.
Academia is currently involved rather late in the traditional drug development model (see below), usually at phase III for larger trials or at phase IV for investigator-driven optimisation or extension of indication after approval, with perhaps some work in target and drug discovery. Everything in between is largely led and organised by pharmaceutical companies, but I think there are opportunities for academics to contribute more in the earlier stages of clinical development.
Research and development costs for a novel oncology compound are more than $1 billion, and the costs are increasing. Despite this high cost, 75% of cancer drugs have no meaningful effect on the patient. The number of clinical trials has declined over the past decade, but costs and failure rates remain high. Why do so many trials fail? Up to 30% of trial sites never recruit a single patient, expending costs and effort for nothing. This could improve with better selection, knowing which centres can really deliver. Over half of the trials do not meet their enrolment targets, so will never give us an answer, which is worse than a negative trial in terms of failure to provide useful information.
 The current model of drug development is not sustainable. It has a high failure rate, the high cost of new drugs results in some people being denied access and, most importantly, large numbers of patients continue to being treated with ineffective or insufficient regimens as a consequence. Drug development needs to change in a way that recognises the far-reaching changes to the landscape of how we conduct trials: with more modern and efficient tools, we can do more, and we can do it more efficiently. The landscape is also changing in how we practise medicine, moving from working in separate medical specialties to interdisciplinary disease management teams using a problem-centred rather than discipline-based approach. Non-specific chemotherapy is being replaced by rational and targeted treatments; organ- and histology-based classifications are moving to an approach that is driven by signalling pathways, which can be used to detect patients at risk and develop more effective drugs with less toxicity. We are also moving from treating disease symptoms and loss of normal function to prevention, intervening before symptoms appear, with the aim of preserving normal function.
The current model of drug development is not sustainable. It has a high failure rate, the high cost of new drugs results in some people being denied access and, most importantly, large numbers of patients continue to being treated with ineffective or insufficient regimens as a consequence. Drug development needs to change in a way that recognises the far-reaching changes to the landscape of how we conduct trials: with more modern and efficient tools, we can do more, and we can do it more efficiently. The landscape is also changing in how we practise medicine, moving from working in separate medical specialties to interdisciplinary disease management teams using a problem-centred rather than discipline-based approach. Non-specific chemotherapy is being replaced by rational and targeted treatments; organ- and histology-based classifications are moving to an approach that is driven by signalling pathways, which can be used to detect patients at risk and develop more effective drugs with less toxicity. We are also moving from treating disease symptoms and loss of normal function to prevention, intervening before symptoms appear, with the aim of preserving normal function.
Towards personalised medicine
The future for more efficient treatment development is to move towards personalised medicine, resulting in the right treatment for each individual patient so they can receive the optimal treatment according to their personal profile, the host profile, and the tumour profile. The move from histology to molecular disease classification results in disease fragmentation from relatively common cancers into many different, rarer subtypes, as illustrated below for lung and breast cancer.
It is important to recognise that expression profiles of thousands of patients are needed to generate a robust gene list that accurately predicts outcomes in cancer, and some current predictors are not as reproducible as we would like to think. Only by bringing a lot of data together can we analyse molecular subtypes accurately and understand what is happening. Analysing thousands of variables in hundreds of samples poses a major challenge, requiring expert support from biostatisticians.
The idea that a targeted treatment blocking a single signalling pathway will be effective is too simplistic because there are multiple redundant signalling pathways, so blocking one means an alternative is then used. We also need to recognise that not every target identified is druggable. In order to identify druggable targets we need companion diagnostics and also standardisation of testing.
We need help from regulators to facilitate new drug development using a personalised medicine approach. Many current regulations that are designed to protect patient safety, stifle patients’ access to innovation. It is important to focus on measurable parameters and define what constitutes a meaningful endpoint, including quality of survival. How low or how high do we want to set the bar for new treatments? Is a median survival prolongation of six weeks worth it? Does the median mean anything? These are all questions that we have to ask and for which answers have to be found individually for each tumour type and treatment we investigate. We need to ensure that trials are representative of the ‘real world’.
Collaboration versus competition
Disease fragmentation is a challenge, but it can be overcome with effective collaboration and sharing of data, including finding a way for competing groups and industries to collaborate where there are synergies. National healthcare systems need to find ways to work together, and different medical disciplines need to collaborate.
Two examples of precompetitive collaboration are the Structural Genomics Consortium (SGC) at the Universities of Oxford and Toronto, supported by private funders and charities, and the Innovative Medicines Initiative (IMI), which brings together the European Commission and the European Federation of Pharmaceutical Industries and Associations. They work in a precompetitive way by carrying out the basic science relevant to drug discovery. This is now done largely in the preclinical field – in imaging and in new technology – but I think we can also find ways for more collaboration, rather than competition, on the clinical side.
The issue of intellectual property (IP) is often a concern in consortium agreements, and can lead to undue delays or good ideas may not be pursued at all, even though it may be more rewarding to have a small share of a successful operation than 100% of a failure.
The changing approach to drug development requires more focus in the early phases to ensure we expose fewer patients to pivotal trials that fail (see figure). We need to ask the right questions upfront in early clinical trials: is the target present and is the target relevant for the tumour being investigated? What is the interaction between the tumour and the stroma? What redundancy of pathways is there? We also need to learn more about the pharmacology and assessing whether the drug reaches the target and if there are any off- target effects. Can we image the drug effect with modern technology?
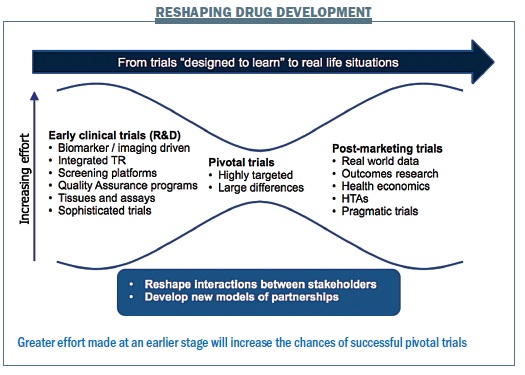 Greater investment in early clinical trials requires that we learn more from every single patient we treat, mandating translational research rather than seeing it as optional. This will ensure we expose the least number of patients to potentially ineffective and toxic agents. Then we will be able to carry out pivotal trials in enriched populations, which will require fewer patients to demonstrate efficacy, because there will be a much greater signal. We can then move on to test a new drug again on a larger scale in the real world, which requires learning how to collect data in a simplified manner. We need to reshape the interaction between the different stakeholders and develop new models of partnerships. Data collection and data sharing is in the interests of each and every patient, and it should be mandatory – overemphasising data protection obstructs progress.
Greater investment in early clinical trials requires that we learn more from every single patient we treat, mandating translational research rather than seeing it as optional. This will ensure we expose the least number of patients to potentially ineffective and toxic agents. Then we will be able to carry out pivotal trials in enriched populations, which will require fewer patients to demonstrate efficacy, because there will be a much greater signal. We can then move on to test a new drug again on a larger scale in the real world, which requires learning how to collect data in a simplified manner. We need to reshape the interaction between the different stakeholders and develop new models of partnerships. Data collection and data sharing is in the interests of each and every patient, and it should be mandatory – overemphasising data protection obstructs progress.
Everybody talks about translational research, however not much is done in practice. One way the EORTC is addressing this is with a molecular screening platform – the Screening Patients for Efficient Clinical Trials Access (SPECTA) platform. This ‘takes the trial to the patient’ by ensuring tumours are subtyped and categorised at a molecular level early on, and then if their cancer recurs after standard treatment upfront they can enter a trial for second- or third-line treatment that fits their characteristics. This requires collaboration between industry and academia to ensure new treatments can be used in appropriate patients. Having an independent molecular screening platform ensures that patients can be directed to appropriate trials.
In this approach the patient’s tumour tissue goes to a biobank upfront where it is analysed in a quality controlled manner, and the information is stored in a clinical database. If the patient progresses, information is exchanged to assess suitability for trials of novel treatments that would be appropriate. The trial may sometimes go to the patient or sometimes the patient will travel to the trial.
The EORTC SPECTA programme tries to categorise different types of cancers. A platform is already up and running in colorectal cancer, with others in advanced development for lung, prostate, brain and melanoma. We are working closely with pathologists, experts in biobanking, biostatisticians, industry, patient representatives and regulators to ensure procedures meet regulatory standards.
Standards and quality assurance are essential to ensure that pathology and molecular testing are done in the same way in different laboratories. Individual academic laboratories alone may not have sufficient numbers of samples to fulfil accreditation and standardisation procedures; however, collaboration with a certified platform may allow them to develop the expertise in a specific area. A centralised platform offers a service that can be much cheaper and more efficient.
The EORTC infrastructure supports new-generation clinical trials. We have quality assurance platforms in radiotherapy, and we have a platform for imaging so that we can analyse and compare images in a unified way.
Where do we have opportunities as academics?
I think there are plenty of opportunities for academics to work, for example, on combination radiotherapy plus drugs and/or radiosensitisers. Progress in the treatment of numerous solid tumours, including brain tumours, head and neck cancer, lung cancer and cervical cancer, over the last twenty years has always been made when we combined drugs and radiation. There are plenty of opportunities to explore new approaches of combined treatments with radiation, but this can only be done when we work in close collaboration among academics and together with industry partners.
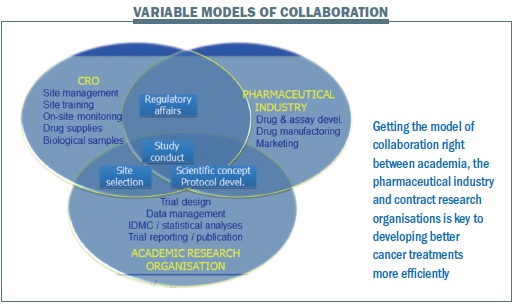
When you consider collaboration – and EORTC is an example of an academic cooperative group – every one of the stakeholders has something to bring to the table. But there is a lot of overlap and things we can do better together. There are variable models of collaboration depending on the project, and depending on the new treatment and approach (see figure below). I think we need to bring academia to the table early on when it comes to the scientific concept, protocol development, site selection, how to carry out trials, and how to do it in the most efficient way.
 What can an academic consortium such as EORTC offer? We offer expertise in clinical development and in specific disease areas that is only available in a highly sophisticated and well-run group with appropriate experience, with members who know each other and who have worked on several previous protocols. This expertise spans decades, not just a few years. Previous experience means academia has expertise in disease benchmarking. Only academia has a long-term horizon: with EORTC, trials have been followed up for up to 50 years. How else would you determine long-term toxicity complications or secondary malignancies?
What can an academic consortium such as EORTC offer? We offer expertise in clinical development and in specific disease areas that is only available in a highly sophisticated and well-run group with appropriate experience, with members who know each other and who have worked on several previous protocols. This expertise spans decades, not just a few years. Previous experience means academia has expertise in disease benchmarking. Only academia has a long-term horizon: with EORTC, trials have been followed up for up to 50 years. How else would you determine long-term toxicity complications or secondary malignancies?
Effective collaborative research needs a network of people who trust each other and who are highly motivated. They need to feel part of something, which ensures they are highly motivated and so perform better than just being ‘hired for service’ in trials run solely by industry. I think independence is important and a platform of academics can be helpful for trials that test several competitors’ compounds and strategies. We can achieve synergies including use of one molecular testing platform, testing for a series of aberrations and then directing patients to the most promising protocol. Trials may even share a common control arm.
Academic consortia such as the EORTC can provide the experience and expertise to get a trial done efficiently, from the initial concept to protocol development and trial enrolment. We are often asked the time to first patient enrolment, but the more important question is the time to enrol the last patient – that’s what matters – and we ensure we get the last patient in efficiently. Time to the first patient is confounded by regulatory issues, delays in contracts, remarks and diverging recommendations by ethics committees. Here inclusion of patient advocates may be helpful.
Denis Lacombe, headquarters director of the European Organisation for Treatment and Research in Cancer (EORTC), Brussels, hosted a live question and answer session
Q: Do you think the ‘pick the winner’ model proposed by acute myeloid leukaemia researcher Alan Burnett [head of the Experimental Cancer Medicine Centre in Cardiff, Wales] is a good platform to develop and assess new drugs for clinical trials?
A: It’s one among many models, and there is not going to be one single approach. It depends on how many compounds and approaches you have to test in parallel and what disease is being studied, but it goes in the right direction. It also depends at what stage of the development we use it. Using the ‘pick the winner’ model with some controlled randomised designs early on and trying to discard the losers very early on would help a lot. We currently have too many losers that we take along too far in the drug development process.
Q: How can we better collaborate to ensure rational combinations of different anticancer agents are used together earlier in the development process, despite competition between different companies?
A: We need to hit several targets at several levels so we need to combine treatments. Doing this in a non-competitive way using a somewhat neutral platform will help. Pharma is used to collaborating in joint ventures and co-developments, so this could be applied to early development perhaps, with two compounds in combination. Currently 56% of trials fail because of lack of efficacy, so we need to overcome this.
Q: We need more international access to patients because of disease fragmentation. How do you see the future of this in Europe, because we do not necessarily operate in an optimal regulatory environment?
A: We need to stand together in Europe otherwise pharmaceutical industry drug development is going to move elsewhere. Our healthcare systems are too fragmented, with a national rather than international focus. Other regions of the world are bringing resources together, picking up in innovation. I would call on the EU and regulators to partner with us in order to develop better treatments to achieve improved health and quality of life for the European population.



Leave a Reply