



Generics markets are gearing up for the expiry of patents on some of the first targeted cancer drugs. Its good news for greater access, but patients want reassurance that switching to generics wont put them at risk.
 Generic drugs are a huge and complex part of the healthcare market. Each year, dozens more become available as the patents that protect exclusive marketing rights for the originator drugs expire or are circumvented, and as developing countries gear up their pharmaceutical sectors.
Generic drugs are a huge and complex part of the healthcare market. Each year, dozens more become available as the patents that protect exclusive marketing rights for the originator drugs expire or are circumvented, and as developing countries gear up their pharmaceutical sectors.
Cancer drugs are no exception. According to recent figures, in a total global oncology drugs market approaching $100 billion, revenues from generics are growing at twice the rate of the market as a whole, and will reach more than $20 billion by 2018. The vast majority of all drug prescriptions are already for generics – more than 80% in the US, for example.
The market is complex for several reasons. One is that rules for marketing exclusivity for medicines, e.g. for orphan drugs (for rare diseases), vary across countries, giving rise to a patchwork of opportunities for generics. Indeed in India there has been a direct challenge to drug patents, the notable case being for imatinib (Glivec). Then there is the economics of producing generics. With prices for some reduced to just a few cents a dose, incentives to remain in production can disappear, which is one of the contributors to well-publicised shortages of cancer drugs in recent years.
And there are concerns about the quality of generic drugs. While most of the small-molecule cancer agents are straightforward to produce, they may be produced in facilities that differ from those of the originator company in levels of quality control and also in the amount of active and other substances used to make up a pill or an injectable dose.
A clear distinction can be made between small-molecule drugs, such as tyrosine kinase inhibitors (TKIs) like imatinib and chemotherapies like cisplatin on the one hand, and large-molecule ‘biologics’, such as monoclonal antibodies, on the other. The latter cannot be copied in the same way as small-molecule agents, and are part of a growing interest and market in ‘biosimilars’, which have more demanding regulatory requirements.
However, there is also a growing category in-between, so-called complex non-biological drugs, which also cannot be subject to conventional generic regulation. A good example in cancer is Doxil, which is a nanotechnology formulation of the chemotherapy drug doxorubicin, and was introduced in 1995. Here the drug is encapsulated in nano-sized liposomes, so assessing the equivalence of a generic version poses challenges concerning the size and behaviour of particles. In 2012, the US regulatory body, the FDA, rapidly approved a generic version of Doxil to ease a shortage of the drug. However, questions about how regulators approve follow-on versions of drugs with novel delivery mechanisms and multiple agents are still the subject of discussion.
Quality control
The vast majority of generic cancer drugs today are small molecules that have been subject to relatively simple regulation for a single active ingredient. For oral drugs, regulators look for evidence that the drug is ‘bioequivalent’ to the original through tests on bioavailability, which is the amount of the drug that reaches the bloodstream – the tests measure the rate and extent of absorption, and bioequivalence is shown by testing both the generic and the original in small groups of people, usually healthy male volunteers. Drugs delivered intravenously are usually exempt from bioequivalence tests, as it is assumed that all the drug is reaching the bloodstream.
Regulators also look for data on the manufacturing process and stability of the drug, to ensure compliance with the general regulations concerning good manufacturing practice (GMP) that apply to any drug. Other than that, a generic small molecule faces far less stringent regulation to reach the market than does the original agent, which will have been required to go through clinical trials to demonstrate good evidence of efficacy and safety in patients with the target disease.
Atholl Johnston, professor of clinical pharmacology at Queen Mary, University of London, says that doctors and patients can be confident that most of the generic drugs that are dispensed in Western nations are of high quality. The manufacturing plants are inspected frequently, he says, and the dossiers submitted to the European Medicines Agency (EMA) and other regulators for approval will tick all the right boxes for bioequivalence and safety factors. This is borne out by a 2009 review of 12 years of bioequivalence data from the FDA of more than 2,000 studies of approved oral drugs, which showed the criteria used have been working well (Ann Pharmacother 2009, 43:1583–97).
“Doctors and patients can be confident that most generic drugs dispense in Western nations are of high quality” “But there have been problems with both branded and generic drugs,” Johnston notes. “Recently there have been several recalls for Tylenol [paracetamol] products in the US, and the FDA has also uncovered many shortcomings in the major Indian generics maker, Ranbaxy, such as signatures on quality documents of people who are on holiday and falsification of drug stability data.” In 2013, Ranbaxy’s US arm had to pay fines and claims totalling $500 million relating to the manufacture and distribution of certain adulterated drugs made at two of Ranbaxy’s manufacturing facilities in India, the US Department of Justice reported.
Johnston adds: “All drug regulation is based on trust and what companies tell you – and if we go looking for problems we will find them.” Harmful contamination may be unusual in products made in or supplied to Western nations, but they can reach patients, such as in the UK where an intravenous feed caused a bacterial infection that killed three premature babies in 2014. In India last year a number of women undergoing sterilisation died after receiving antibiotics contaminated with rat poison – developing countries face greater risks from lower standards, but many generics that are used in the West are made in plants in countries such as India.
Impurities, excipients (the many different substances such as preservatives and coatings that the active ingredient is packaged with in a pill or vial), and the amount of active agent itself can all have an impact on the efficacy and side-effects of drugs, adds Johnston, and the approval of oral generics mainly depends on meeting an acceptable range of activity compared with the original drug, not an exact match. Dossiers submitted to the EMA do have to disclose impurities, and the EMA also asks for ‘pharmacovigilance’ follow-up of generics, but globally there are many generic drugs for which companies offer little or no support and monitoring, and regulators don’t require generics to undergo phase IV post-marketing studies.
“Globally there are many generic drugs for which companies offer little or no support and monitoring”
A number of classes of drugs, including those used in oncology, have a narrow therapeutic window (a narrow dose range where the drug is effective but not too toxic). The potential variation in a generic could therefore result in a patient receiving a less than optimum dose – or too much. There are few studies on variation in more than a few generic cancer drugs, but Johnston points to one carried out by a French team that compared the quality of generic formulations of docetaxel available in emerging countries – docetaxel is a chemotherapy drug widely used in a number of tumours including metastatic breast cancer (proprietary name Taxotere, from Sanofi).
Wide variations
The researchers acquired 31 versions of docetaxel in 14 countries including Brazil, China, Egypt, India and Vietnam, demonstrating the large number of generics that can be available for one agent (at the time of the study generic docetaxel was not yet available in the US, Japan or Europe as it was still on-patent). Using Taxotere as the reference, they found that 21 generics contained less than 90% docetaxel, 11 of which had less than 80% of what would be expected. Only 10 were in the acceptable range of 90–110%. They also measured impurities, setting a conservative limit of 3% (the reference was 1.6%). They found that 23 of the generics had impurities levels of more than 3%, and many of the impurities were not detected at all in the reference drug, although this study could not identify what these substances were (see Curr Med Res Opin 2008, 24:2019–33).
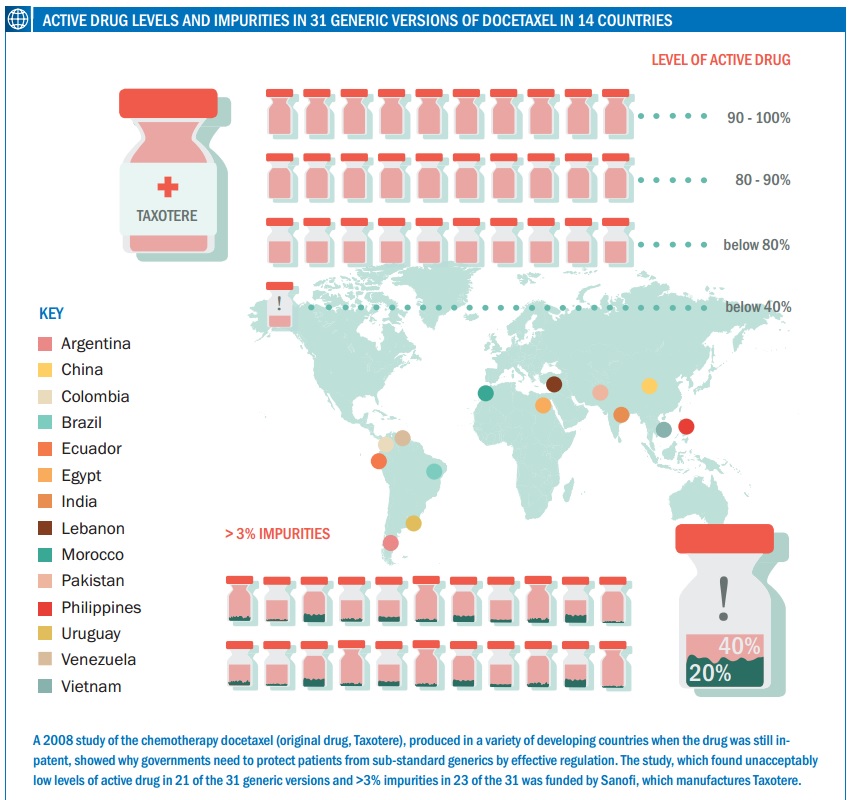 One of the generics, from India, had a docetaxel content of less than 40% of the reference drug and a 20% level of impurities, showing just how poor they can be. The authors conclude: “The number of generic docetaxel formulations failing to meet internationally recognised quality criteria is a concern, in particular given the potential clinical consequences of patients receiving a lower dose of docetaxel than expected.” They also note that the findings were in line with studies of other types of generic drugs, such as for ciprofloxacin and clarithromycin (both antibiotics) and clopidogrel (which prevents blood clotting).
One of the generics, from India, had a docetaxel content of less than 40% of the reference drug and a 20% level of impurities, showing just how poor they can be. The authors conclude: “The number of generic docetaxel formulations failing to meet internationally recognised quality criteria is a concern, in particular given the potential clinical consequences of patients receiving a lower dose of docetaxel than expected.” They also note that the findings were in line with studies of other types of generic drugs, such as for ciprofloxacin and clarithromycin (both antibiotics) and clopidogrel (which prevents blood clotting).
One generic had a docetaxel content of less than 40% of the reference drug and a 20% level of impurities
Since the study was published, docetaxel has come off patent, and the quality in emerging markets may have improved, and different products will be available, but it shows what can happen in countries with poor regulation, where a generic drug may border on being counterfeit. It’s hard to get a picture of the market because, although countries should have their own database of authorised agents, there is no public global database of generic drugs that could make their way into various supply chains, often rebranded by other suppliers.
A recent clinical study on docetaxel, carried out in Canada on a population of breast cancer patients, compared just one generic with the original. It found little difference in adverse events, bar quite a large increase in grade IV febrile neutropenia in the generic group (see Ann Pharmacother 2014, 48:447–455). But this drug is likely to be of high quality, given it is on the Canadian market, which is said to have one of the world’s best bioequivalence inspection regimes.
Also last year, the FDA issued a warning about an adverse effect from docetaxel. The drug is administered in a solvent containing ethanol (alcohol) and has been issued in two vials, one containing the solvent, that are mixed before administration. In 2009, Sanofi introduced a single-formulation already prepared with alcohol, which had the effect of doubling the alcohol content over the two-vial preparation; other generic suppliers of a single-vial docetaxel can have even more – Pfizer’s preparation has over three times more.
This might seem relatively trivial, but the FDA references a letter by two oncologists in the UK who found a male patient receiving palliative care showed symptoms of alcohol intoxication with the new formulation. They point out that alcohol behaves differently when injected rather than drunk, and in cases such as their patient it could render him unfit to drive, which may have led to him rejecting further chemotherapy, and for this patient there was also “a real risk of a relapse in alcoholism”. People may also have religious objections.
In Australia, Pfizer’s docetaxel application was withdrawn before a final decision by the country’s regulator, but it was about to be refused owing to both alcohol and propylene glycol content, and concerns about bioequivalence – in this case the regulator seemed to want a human study despite it being an intravenous drug (Pfizer did submit a study carried out on dogs). In contrast, the UK, which acted as a reference member for most EU countries, and the US have both authorised this generic.
For Johnston, this is just one of many examples of the difficulties that patients can face when they are switched to a new formulation or generic, often without warning or appreciation of differences that, while not necessarily intrinsically harmful, could have a significant impact, for instance on adherence. Healthcare providers and pharmacists are often able to switch prescriptions between a number of generics, and patients only find out if they look closely at the packaging.
A primary motivation to dispense generics is of course cost, but Johnston says that if there are problems with drugs the savings could be wiped out. He has a particular interest in transplant drugs where, as he points out, the cost of rejection episodes and of losing a transplanted kidney owing to a poor drug could be great. “You have to ask whether a drug is mission critical – if so it’s a nonsense to keep switching among a number of generics as you just can’t do effective pharmacovigilance then.”
“You can’t do effective pharmacovigilance if you keep switching among a number of generics”
Patients want harmonisation and transparency
A group that is keeping a close watch on ‘mission critical’ cancer generics is the CML Advocates Network, which unites chronic myeloid leukaemia patient groups across the world. It has a dedicated section for generics on its website and has organised several sessions at conferences. As with so many other issues in oncology, it is the drug imatinib – the first TKI – that is the main focus. Imatinib has been copied extensively, and the network is surveying these generics, presenting the results in an ‘unofficial’ CML TKI register. Currently there are more than 60 imatinib and dasatinib generics – dasatinib (Sprycel) is another TKI used in first-line CML treatment – and there will more to come for various cancers, as other drugs come off patent or are even compulsorily licensed for generics.
In 2012, India compulsorily licensed one drug – Nexavar (sorafenib), a cancer drug – giving a generic maker (Natco Pharma) a licence to make it, as Natco argued that the patent holder, Bayer, had not adequately introduced the drug in India. In another high-profile case, India rejected a claim by Novartis for exclusive marketing of its formulation of imatinib, as generic versions were already on the market because the country did not recognise the patent system until 2005, and Novartis’ imatinib also did not meet the requirement of ‘enhanced therapeutic efficacy’. Currently, imatinib is made by ten generics companies in India.
Šarūnas Narbutas, who is a CML patient active in the advocates network and in cancer advocacy in his home country, Lithuania, says there is concern that the molecular targeting of drugs such as imatinib poses particular challenges for assessing their efficacy and safety. “We just don’t have the long-term data about possible effects of different excipients and stability of these drugs, and in any case generic companies must use an alpha form of the imatinib crystal, at least until the patent expiration in 2019, whereas Novartis, the marketing authorisation holder for Glivec, uses the beta form. Legally, they are ‘bioequivalent’ but they can produce different side-effects and clinical outcomes.”
The molecular targeting of drugs such as imatinib poses particular challenges for assessing their efficacy and safety
Imatinib has been a spectacular success with CML, but it is a strong candidate for therapeutic drug monitoring to ensure that patients attain and maintain response, and Narbutas says people are rightly worried about putting their response at even the slightest risk if they are switched to a generic. “Before imatinib generics came to Europe we had case reports from India that some patients who were switched to substandard versions had severe side-effects or lost response, and there have been conflicting studies since from Colombia, Egypt, Iran and Turkey.”
He notes that one of the largest ongoing studies so far is in Serbia, where all existing and new patients, about 220 people, were put on a generic imatinib called Anzovip without warning in 2012, much to the alarm of advocates in the country. Although supplied by a local company, the suspicion was that it is repackaged from an Indian provider, and it is much cheaper. But so far, concerns about its efficacy and safety have been unfounded, report Serbian haematologists, who are monitoring their patients closely. A few long-term patients did lose cytogenetic response and were switched to nilotinib (Tasigna), but most patients followed up so far have seen equivalent responses to Glivec and no different toxicity after 18 months. Another year’s data is needed, Narbutas adds, but ideally Serbia’s CML patients would have liked to have had the drug independently tested by the government at a European laboratory.
“There are similar issues with TKIs for other cancers, and also for biosimilars,” says Narbutas. “You need to be closely followed up to see whether you are responding in the same way as if you were taking the originator drug, and we strongly advocate not to keep jumping between generics; you should stick to one generic for at least a year to allow for comparable medical history in each patient.” The key point is that advocates are not against generics given that they open up access to key drugs, but there needs to be much more transparency and information about their use, and effective regulation on efficacy and safety. CML advocates in Canada note that patients can be switched to a generic by pharmacists and their oncologists may not know unless the patient tells them. A recent review of generic imatinib in Canada and the EU does provide reassurance, finding there is no evidence of less effectiveness (J Oncol Pharm Pract 2015, 21:76–79).
Advocates are not against generics, but there must be much more transparency and information about their use And it is not just poorer nations that can benefit from generic cancer drugs. A recent study in the US found that women taking hormone therapy for receptor-positive breast cancer are more likely to continue treatment on generics owing to lower out-of-pocket costs.
There is research at stake too, as cancer generics are increasingly in demand not only for standard treatments but also for clinical trials. In a number of cases the original marketing authorisation holder may not even be in the market anyway. This places more responsibility on the major generics firms to provide education and support for their products, and transparency about quality processes. Johnston mentions a South African generics firm that rejected an approach to produce a drug under contract, when it was asked to cut the number of quality control steps it takes – there can be 40 or more such steps. Narbutas adds that advocates who have been invited to see how Glivec is made by Novartis were impressed by the extent of the quality control, and inevitably this raises questions about smaller companies with fewer resources.
But the market remains confusing, and there are no guidelines for patients about the issues arising from switching to and among generics. While the world’s drug regulators are mostly in agreement about the bioequivalence approaches used to assess generic drugs – there are more similarities than differences, as a review found (AAPS Journal 2013, 15: 974–990) – patient groups want more regulatory harmonisation.

www.cmladvocates.net/133-generics/354
Signs of progress
Two recent initiatives may help. In 2012, the International Generic Drug Regulators Pilot (IGDRP) was launched to develop a more global picture, and recently the European Union has announced it will lead a project within this using the EU’s decentralised procedure “as a model to accelerate the assessment of applications for generic medicines”, one of the pilot’s work packages.
Then in 2013, the EMA, FDA and some EU member states said they would start sharing information on inspections of bioequivalence studies submitted to them – a move that advocates had pushed for at a meeting at an ASCO conference, according to Narbutas. The collaboration also includes inspections of facilities where the studies are carried out.
It is early days for these projects and India is notable for its absence so far from the international pilot, and is not yet an observer at the longer standing ICH (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use), which is led by Europe, the US and Japan.
However, the recent focus on generics at international level is encouraging, though an overriding issue is that countries may have to pay more for high-quality generics rather than driving the prices down to levels where some companies will cut corners.


Leave a Reply