Sarcomas and gastrointestinal stromal tumours include a wide variety of biologically diverse cancers, many of them very rare. Paolo Casali, a leading expert, presents an update of the latest evidence on the best way to manage them.
This is an edited version of a presentation by Paolo Casali, from the National Cancer Institute, Milan, Italy, that was first transmitted to the 2nd ESO–ESMO Latin-American Masterclass in Clinical Oncology for the European School of Oncology. It is edited by Susan Mayor.
Sarcomas and gastro-intestinal stromal tumours (GISTs) are rare cancers. The incidence of soft tissue sarcomas in adults is 4.5 per 100,000 population per year, and for GIST it is 1.5 per 100,000 population per year. Osteosarcoma has an incidence of 0.3, Ewing’s sarcoma 0.2, and rhabdomyosarcoma (RMS) 0.1 per 100,000 per year.
The very low incidence of these rare cancers contrasts with an incidence rate of 300 per 100,000 per year for benign tumours. This poses a challenge for clinicians and inevitably results in delays in diagnosis. This is true for a range of soft tissue lesions, such as uterine leiomyosarcomas, whose benign counterpart – uterine leiomyomas (fibroids) – is one of the most common conditions in women.
Treating localised tumours
Surgery is the standard approach for treating localised adult soft tissue sarcomas. Radiation therapy is used quite frequently, although possibly less frequently in current practice. However, when soft tissue sarcomas are high grade, deeply located and require surgery that is not compartmental surgery, then radiation therapy is indicated in principle. Use of adjuvant doxorubicin plus ifosfamide is not standard treatment, although it can be used on an individualised basis.
Treating advanced disease
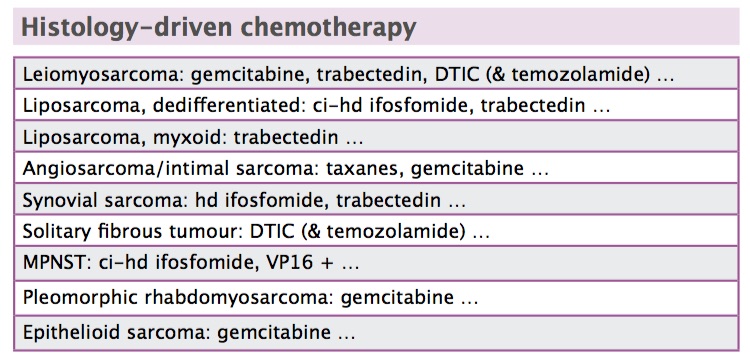
Surgery is standard treatment for isolated metastatic disease to the lung. Standard chemotherapy is doxorubicin, although doxorubicin plus ifosfamide is widely used, depending on a patient’s presentation. Histology-driven chemotherapy may be a choice as second- or later line therapy.
Dedifferentiated liposarcoma
Continuous-infusion high-dose ifosfamide has been shown to be active in dedifferentiated liposarcoma
Surgery for lung metastases may be the used when lesions are isolated in the lungs. The indication for surgery is higher in patients where the previous disease-free interval was long and the number of lesions reasonably low. These are the best prognostic factors. However, there are a variety of clinical presentations, so it is always questionable as to whether to resort to surgery or not.
Myxoid liposarcomas
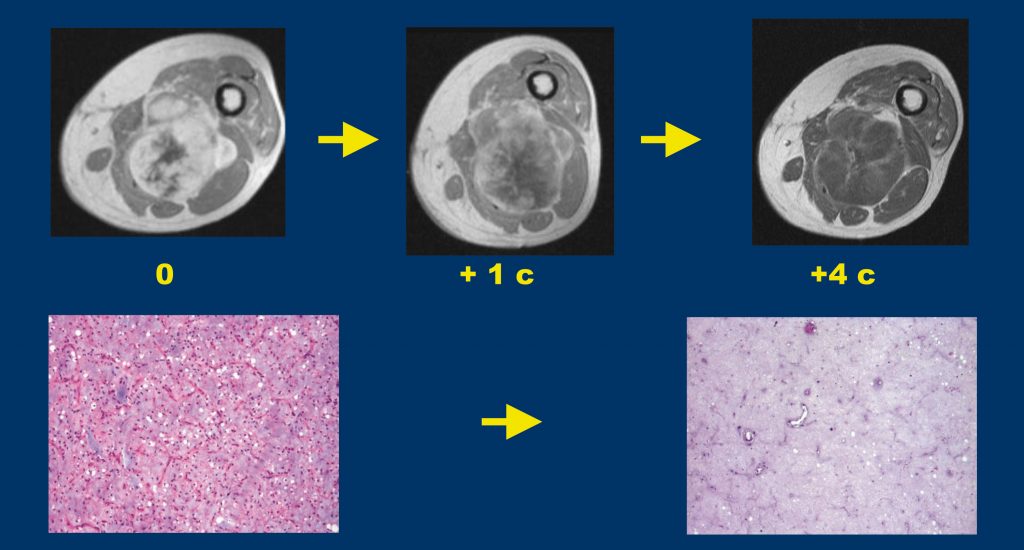
Trabectedin is approved for treating soft tissue sarcomas and has been shown to induce a strong pathological response in myxoid liposarcomas (upper ) and in some cases also tumour shrinkage (lower)
We are inclined to use chemotherapy in addition to surgery in cases where prognostic factors are favourable. There is no evidence behind this, but it seems logical. The question as to whether or not to add chemotherapy to surgery has not yet been settled by clinical studies. We generally give chemotherapy before surgery in these cases, to help ascertain if the patient is responsive to chemotherapy in order to tailor the overall strategy.
The choice of chemotherapy in advanced disease
Doxorubicin plus ifosfamide. A randomised trial carried out by the EORTC Soft Tissue and Bone Sarcoma Group compared the standard treatment of doxorubicin (75mg/m2) alone with doxorubicin (75mg/m2) plus ifosfamide (7.5g/m2). The results showed a difference in progression-free survival in favour of the combination; however, the difference in overall survival was not statistically significant (Lancet Oncol 2014, 15:415). This means that single-agent doxorubicin could still be regarded as the standard treatment. Depending on the clinical presentation, the combination of doxorubicin plus ifosfamide may be used, particularly if it is believed that a tumour response could be useful.
This randomised trial put together all histologies, but the question is how far we need to take account of the complexity of the histology of soft tissue sarcomas. This is a particular challenge for managing the disease and also for clinical trials. The more you put the different histologies together, the less likely you are to see differences that may apply only to some histologies and not others.
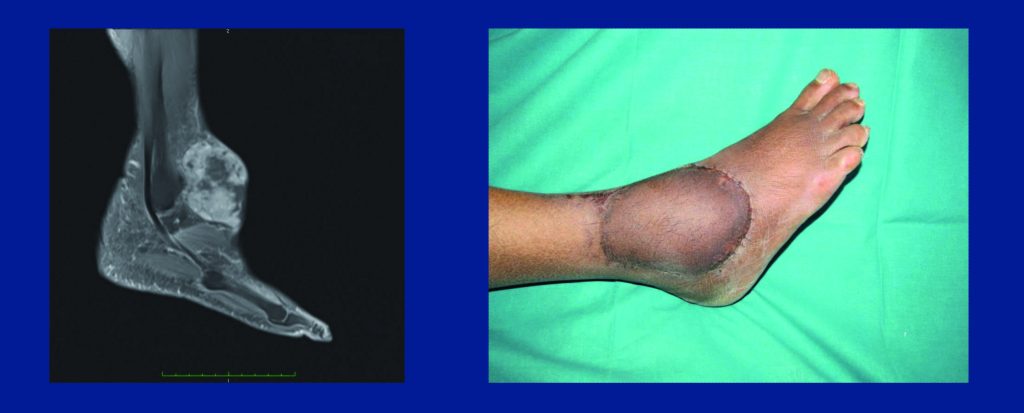
Ifosfamide. In terms of the main drugs used in soft tissue sarcomas, ifosfamide is not active in leiomyosarcomas according to retrospective evidence (JCO 2007, 25:3144–50). As a result, many institutions do not use ifosfamide in leiomyosarcomas. In contrast, ifosfamide is very active in synovial sarcoma. It is also active given as a continuous high-dose infusion over 14 days in dedifferentiated liposarcoma (see figure opposite, top). Two retrospective series suggest efficacy of high-dose continuous infusion in dedifferentiated liposarcomas (Sarcoma 2013, doi.org/10.1155/2013/868973; Clin Sarcoma Res 2014, 4:16).
Trabectedin. Trabectedin is another drug that is now approved in the US and Europe for soft tissue sarcomas. This agent is active in dedifferentiated liposarcoma, but probably primarily in those with less aggressive behaviour. It is especially active in myxoid liposarcomas, more so than in dedifferentiated liposarcoma, with tumour shrinkage in some cases and very convincing pathological response – despite lack of tumour shrinkage – in other cases (see figure opposite, bottom). This may be because trabectedin has a targeted mechanism of action in myxoid liposarcoma, in which the drug displaces the fusion transcript specific to this type of tumour from target genes and promotes a type of differentiation (Mol Cancer Therap 2009, 8:449).
Trabectedin is active in metastatic liposarcoma and leiomyosarcoma, after failure of conventional chemo-therapy (JCO 2015, doi: 10.1200/JCO.2015.62.4734). This includes uterine leiomyosarcomas (Gynecol Oncol 2011; 123: 553–56).
Eribulin. More recently there has been some evidence to show eribulin may be active in liposarcoma, providing an increase of seven months in overall survival compared to dacarbazine (Lancet 2016, 387:1629–37)). However, it is not clear why there is less improvement in progression-free survival. This drug has now been approved by the US regulators, the FDA, and has received a positive opinion from the Committee for Human Medicinal Products of the European regulators, the EMA.
Dacarbazine. Dacarbazine is active in leiomyosarcoma, but much less so in liposarcoma.
Gemcitabine. Gemcitabine is active in leiomyosarcomas although not in any other soft tissue sarcomas, with the exception of angiosarcoma. One study has shown improved progression-free survival with gemcitabine plus docetaxel compared to gemcitabine alone (JCO 2007, 25: 2755–63), but other studies did not confirm this. An option, which we follow at our institution, is to use gemcitabine alone in leiomyosarcomas. This is much better tolerated.
Pazopanib. The antiangiogenic drug pazopanib has demonstrated improved progression-free survival in a phase III study of all soft tissue sarcomas, with the exception of liposarcomas, which were not included (Lancet 2012, 378:1879). Some histologies, including uterine leiomyosarcomas and synovial sarcomas, are more responsive. However, the rebound effect that can occur with antiangiogenic agents may limit the use of pazopanib.
In summary, in some histologies of soft tissue sarcomas, such as leiomyosarcoma, it is possible to use several chemotherapy drugs. Some of these drugs can be used for a relatively long time. Other histologies, such as undifferentiated pleomorphic sarcoma, have far fewer options for medical therapy.
Rarer histologies
Certain rarer histologies may respond to specific drugs:
Angiosarcoma responds to taxanes, which have no effect on other soft tissue sarcomas, at least as single agents. Angiosarcoma also responds to gemcitabine.
Low-grade endometrial stromal sarcomas respond to hormonal therapy with progestins or aromatase inhibitors, but not to tamoxifen, because it is an agonist. The evidence is only anecdotal, but this type of sarcoma is very rare. This type of sarcoma has a non-random chromosomal change. High-grade endometrial stromal sarcoma has a different chromosomal change. Undifferentiated endometrial sarcoma, another type, is very aggressive and not responsive to hormonal therapy.
Some sarcomas have many more treatment options than others
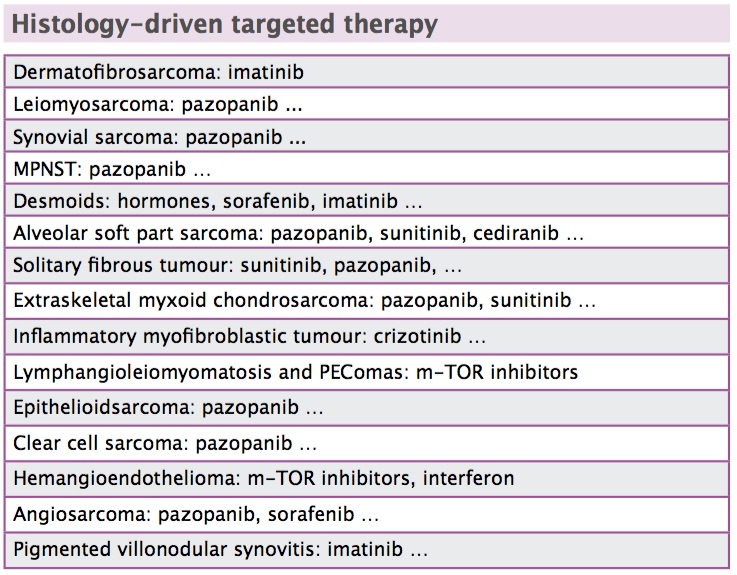
Desmoid tumours may be responsive to hormonal therapy, but their natural history is very erratic, sometimes progressing and sometimes spontaneously regressing. We are using less and less surgery in desmoid tumours and have published a consensus-based algorithm of drug options (Ann Oncol 2014, 25:578–83). Different histologies respond to different drugs. For example, dermatofibrosarcoma has a chromosomal translocation, with an overproduction of PDGF-beta, and responds to imatinib.
This underlines the importance of histology in treating soft tissue sarcomas, with different histologies responding to different chemotherapies and targeted drugs (see tables above). The range of different histologies makes it difficult to carry out randomised controlled trials in soft tissue sarcomas. Results from the trials that are carried out need to be interpreted cautiously, because putting different soft tissue sarcomas together makes it difficult to make sense of the findings.
Adjuvant chemotherapy
A systematic meta-analysis suggested a 10% advantage with adjuvant chemotherapy in high-risk, localised, resectable soft-tissue sarcomas (Cancer 2008, 113:573). However, the studies included showed divergent results, with some being negative and others positive.
DTIC dacarbazine, ci – continuous infusion, hd – high-dose, MPNST – malignant peripheral nerve sheath tumours
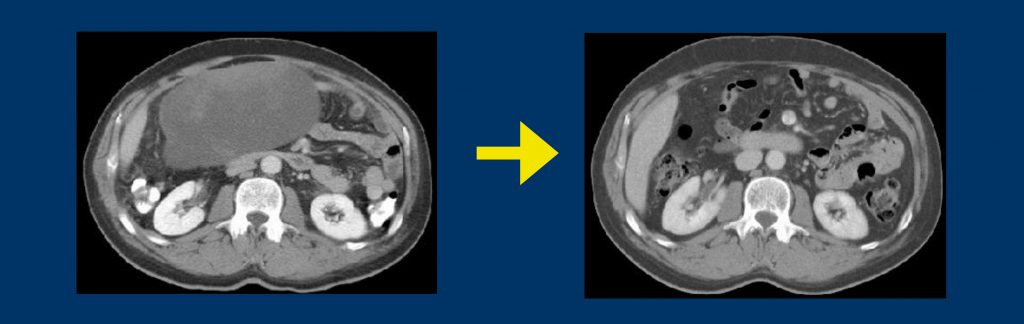
A study in our centre compared three cycles of epidoxorubicin, followed by surgery with or without radiotherapy, with the same regimen followed by a further two cycles of epidoxorubicin in patients with high-grade spindle-cell sarcoma. Results were the same, so we currently propose three cycles of chemotherapy to patients at high risk, quite often before surgery. This is because surgery in patients with high-risk sarcomas can be quite challenging and may potentially involve reconstructive surgery, so it may be better to give chemoradiotherapy before surgery and not after (see figure opposite, top).
We developed an app (Sarculator, see www.sarculator.com) to assess prognosis in patients with soft tissue sarcoma, based on our series, which may be helpful to make treatment decisions on chemotherapy.
MPNST – malignant peripheral nerve sheath tumours
Gastrointestinal stromal tumours (GIST)
Treatment of GIST is quite simple, with standard treatment being surgery. Imatinib is used as adjuvant treatment in high-risk GIST. For advanced disease, imatinib, sunitinib and regorafenib are the three molecularly targeted agents available as first-, second- and third-line treatment. Surgery for metastasis is not standard, but can be used in some cases, even though we do not have convincing evidence for this in addition to medical therapy.
The case for neoadjuvant treatment
Chemotherapy is sometimes used before surgery in patients with high-risk sarcomas, as they can be quite challenging and may potentially involve reconstructive surgery
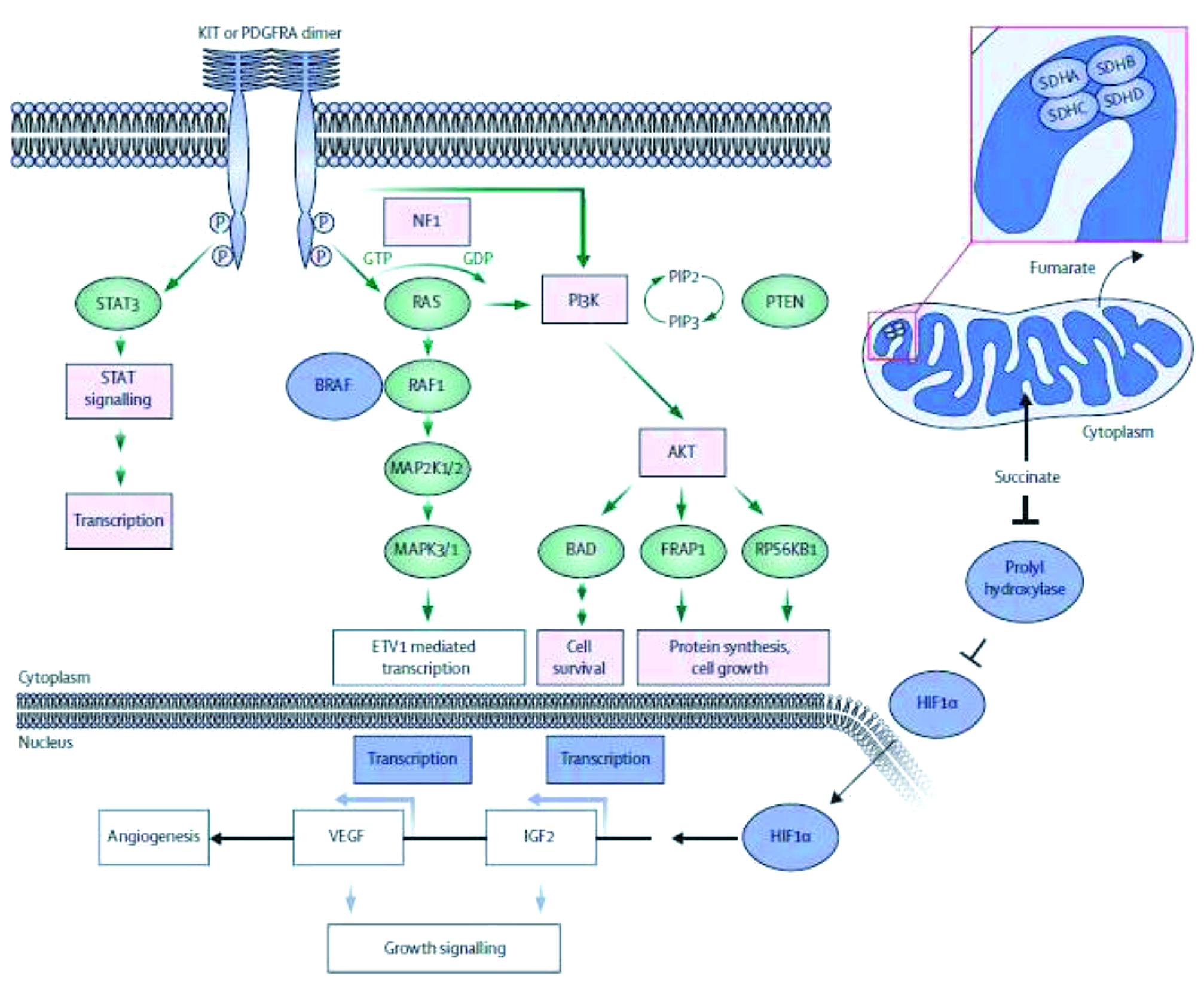
This treatment approach is based on the molecular biology of GIST, with molecular analysis being essential to make decisions on medical therapy (see figure below). Not all GIST responds to imatinib. Exon 11-mutated KIT, which is the most common mutation (60%), responds, but GISTs with the exon 9 mutation may require 800 mg or 400 mg doses of imatinib. The exon 18 D842V mutation is completely insensitive to imatinib. The 10% of GIST cases that are wild type have a completely different natural history and do not respond, in practice, to imatinib, although they may respond to sunitinib or regorafenib. These are divided into two main groups: succinate dehydrogenase- (SDH-) negative and SDH-positive.
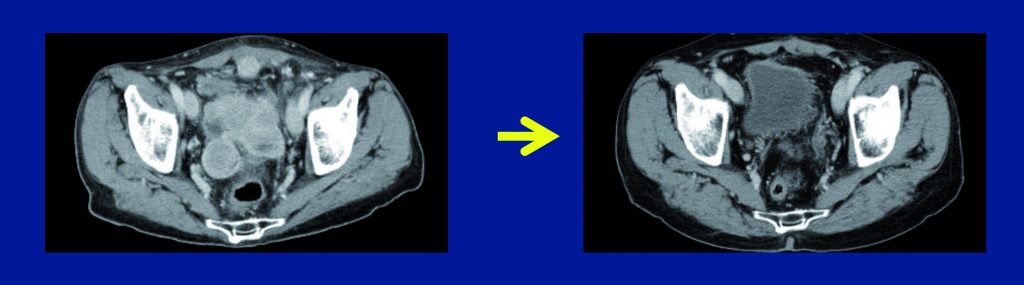
In unresectable or metastatic GIST expressing KIT, imatinib gives a median progression-free survival of two years and a median overall survival of five years (JCO 2008, 26:626–32). Some patients show much longer progression-free survival; however, we do not know who they are, whether they are just the tail of the curve, or whether there are specific reasons why they survive for so long. There are speculations that some kind of immune response may play a role in these patients, with imatinib potentiating antitumour T cell responses through the inhibition of IDO (Nature Med 2011, 17:1094).
Sunitinib demonstrates prolonged progression-free survival and overall survival in patients with advanced GIST after failure of imatinib (Lancet 2006, 368:1329), and regorafenib is also active as third-line therapy, with a benefit of some months compared to placebo (Lancet 2013, 381:295).
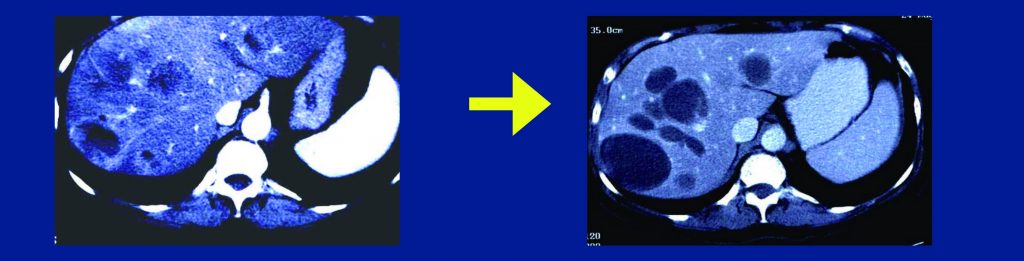
Medical therapy with these agents should be continued long term, otherwise patients lose response. It may be assumed there is not complete pathologic response to imatinib. Radiologically, the response pattern shows typical hypodensity, although there may not be tumour shrinkage. Secondary resistance is the limiting factor, with the main mechanism being molecular heterogeneity. Biopsies show different secondary mutations, in a similar way to other tumour types. Although available drugs show activity against different secondary mutations, this is not helpful in predicting response, because of the heterogeneity.
Focal progression can occur in GIST, so surgery may be an option in patients who progress on imatinib, essentially to prolong survival, as it will not cure metastatic GIST.
Radiological response of GIST to imatinib
Surgery of widely progressing disease is not effective, but surgery for focal progression may provide some benefit. Restarting imatinib can prolong progression-free survival in cases where there are no other drug options to use after failure of imatinib and sunitinib (Lancet Oncol 2013, 14:1175).
Risk stratification in GIST
Source: M Miettinen (2006) Semin Diagn Pathol 23:70
Adjuvant imatinib is effective after resection of localised GIST (Lancet 2009, 374:1097). Longer duration treatment improves survival, with one year better than no adjuvant imatinib, two years better than one, and three years better than two. However, the cure rate is not increased. The benefit of longer-term imatinib is currently unclear, but results from the PERSIST study, which investigated five years of treatment, are awaited.
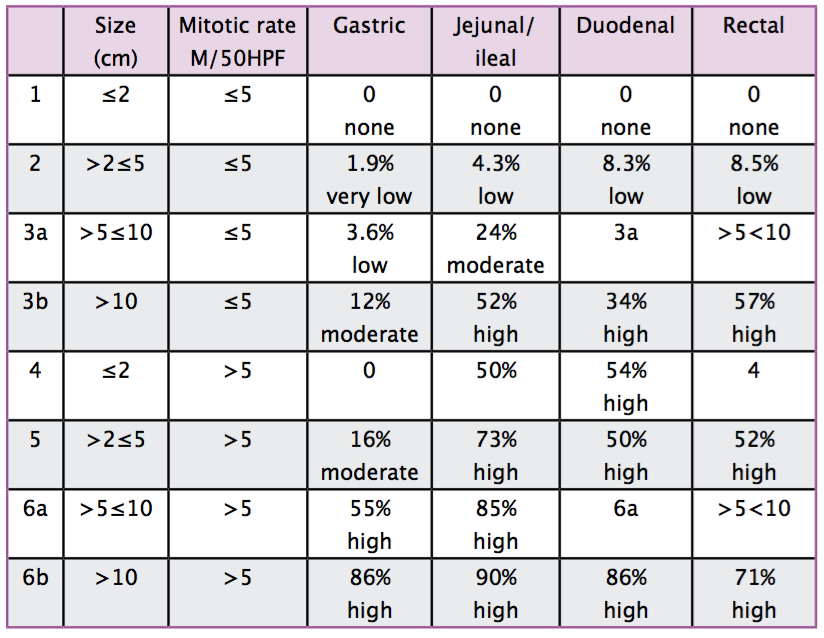
The decision to use adjuvant imatinib should be made jointly with the patient, depending on their risk and the molecular biology of their GIST. The risk assessment should be based on the tumour size, mitotic rate and tumour site (see Table 3). An additional factor to take into account is whether tumour rupture has occurred, because this is a very adverse prognostic factor.
Revising the guidelines
We are currently updating the European Society for Medical Oncology’s Clinical Practice Guidelines for the diagnosis, treatment and follow-up of GIST and soft tissue sarcomas, which will incorporate the research reviewed in this grandround.
Forthcoming educational events
Update on rare adult solid cancers, 25–27 November 2016, Milan
This is the first of an annual event organised by the European School of Oncology (ESO), in association with the European Society for Medical Oncology (ESMO) and Rare Cancers Europe.
Postgraduate Master’s degree in rare cancers
ESO is planning to provide an individualised educational pathway for young oncologists to develop their careers in rare adult solid cancers in collaboration with the Università Degli Studi di Milano and the Fondazione IRCCS Istituto Nazionale dei Tumori. For more information contact: raretumours@eso.net




This is an edited version of a presentation by Paolo Casali, from the National Cancer Institute, Milan, Italy, that was first transmitted to the 2nd ESO–ESMO Latin-American Masterclass in Clinical Oncology for the European School of Oncology. It is edited by Susan Mayor.
 Angiosarcoma responds to taxanes, which have no effect on other soft tissue sarcomas, at least as single agents. Angiosarcoma also responds to gemcitabine.
Angiosarcoma responds to taxanes, which have no effect on other soft tissue sarcomas, at least as single agents. Angiosarcoma also responds to gemcitabine.



Leave a Reply