
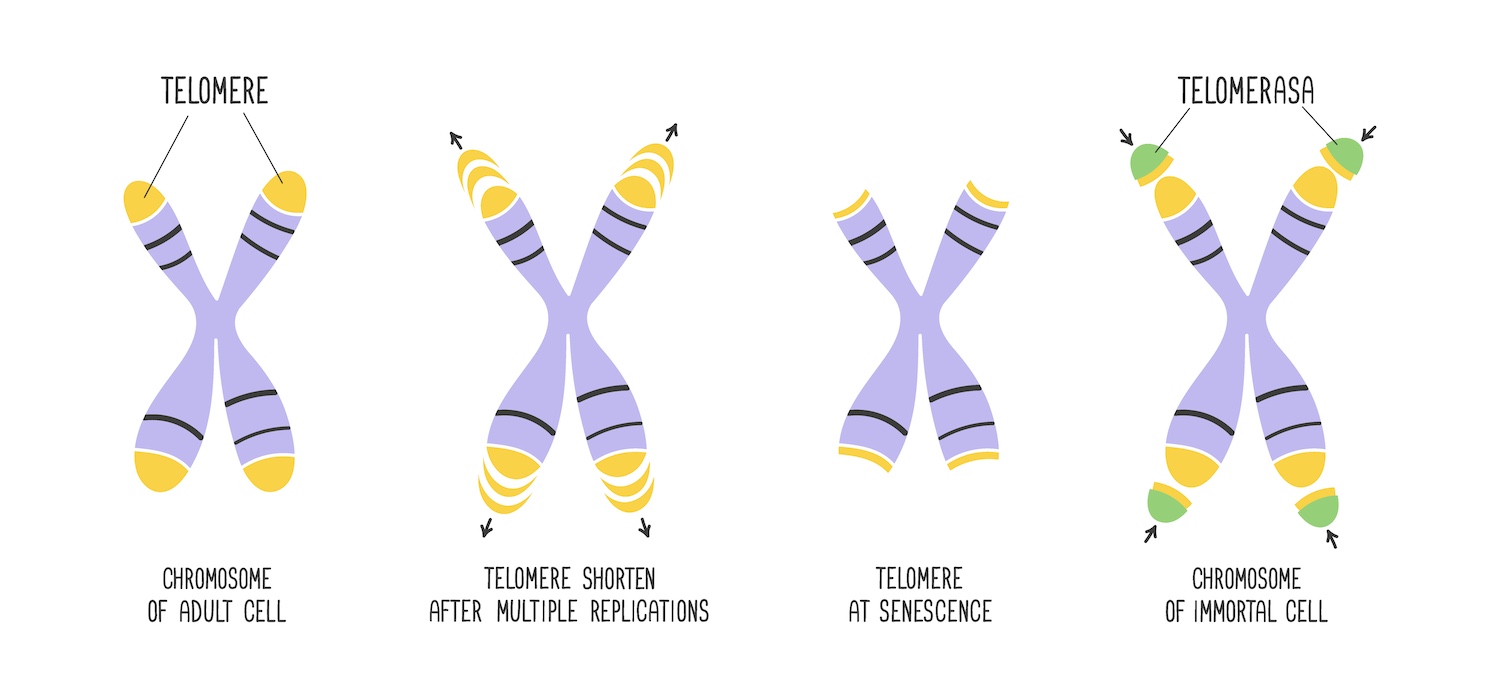
Chromosomes are essential structures in the nucleus of eukaryotic cells that carry genetic information as DNA. DNA is organised into genes, which provide the instructions for building and maintaining the organism. During cell division, chromosomes ensure that each daughter cell receives an accurate and complete set of genetic information. Specialised structures, called telomeres, protect the chromosome ends from deterioration and fusion with neighbouring chromosomes. The existence of these chromosome ‘caps’ was independently hypothesised as far back as the 1930s by Hermann Joseph Muller and Barbara McClintock.
The term ‘telomere’, coined by Muller, derives from the Ancient Greek words for ‘end’ and ‘part’. Telomeres consist of sequences of DNA, specifically the sequence TTAGGG in humans, which are repeated thousands of times. An important structural feature of telomeres is that one of the DNA strands extends beyond the other, creating a single-stranded overhang. This protrusion plays a crucial role in the protective and functional properties of telomeres, contributing to their ability to safeguard the chromosome ends and maintain genomic stability.
It is crucial that the ends of chromosomes, which resemble DNA breaks, do not activate the DNA damage response
If a DNA break occurs in part of a chromosome, spontaneously or due to chemotherapy, the cell’s DNA damage response machinery detects it and initiates repair processes. The cell halts division and focuses on repairing the lesion. If the operation is successful, the cell resumes its normal functions. If the break is irreparable, the cell triggers apoptosis, or programmed cell death. This self-destruction is essential to prevent the proliferation of cells with damaged DNA, thereby maintaining genetic stability and reducing the risk of cancer. However, it is crucial that the ends of chromosomes, which resemble DNA breaks, do not activate the DNA damage response, as this would lead to severe genomic instability and disruption of cellular processes.
Telomeres achieve the silencing of the repair system in a remarkable way, which was discovered twenty years ago by De Lange and colleagues (see box) through studying the proteins that specifically bind to the telomeric DNA. This protein complex, which they named shelterin, as in ‘to shelter’, ‘to protect’, orchestrates the formation of a unique structure – the t-loop. Shelterin (also called telosome) takes the DNA of the telomere and loops it back, then tucks the single-stranded overhang into the telomeric DNA. It looks like a buttonhole, an eyelet (see figure). The cell does not recognise the presence of a DNA end, thus preventing the activation of DNA damage response. The shelterin complex consists of six distinct subunits – TRF1, TRF2, TIN2, Rap1, TPP1, and POT1. TRF2 and TRF1 have DNA remodelling activities relevant to t-loop formation, while POT1 binds and protects the single-stranded overhang, facilitating its tucking into the t-loop structure.
Titia de Lange
Titia de Lange is Leon Hess Professor, head of the Laboratory of Cell Biology and Genetics, and director of the Anderson Center for Cancer Research at The Rockefeller University in New York. Her lab focuses on telomeres, protective elements at the ends of chromosomes that are critical for genome integrity and shorten with cell division. De Lange seeks to understand how telomeres are protected from the DNA damage response by a protein complex called shelterin, how they are replicated and maintained, and how telomere shortening contributes to tumour suppression and genome instability in cancer.
When a cell divides, the DNA replication machinery cannot fully replicate the ends of chromosomes, leading to a progressive loss of telomeric DNA with each cell division. This phenomenon is known as telomeric attrition. After an average of 50 cell divisions, the telomeres become too short to form the protective cap at the chromosome ends. Detecting this as DNA damage, the cell’s surveillance mechanisms trigger the DNA damage response pathways, inducing cell cycle arrest. The limit on the replicative potential of normal cells is known as the ‘Hayflick limit’. It was first discovered in the 1960s by Leonard Hayflick, who demonstrated that normal human cells cultured in the laboratory have a finite replicative lifespan, after which they enter a state of senescence and stop dividing.

Source: Ľ Tomáška, A J Cesare , T M AlTurki , J D Griffith (2020) Twenty years of t-loops: A case study for the importance of collaboration in molecular biology. DNA Repair Republished under creative commons licence CC BY-NC-ND 4
This seminal discovery ushered in a new era of research into cellular aging and senescence in a wide range of research fields. While telomere biology plays a role in cellular aging, the relationship between telomere length and overall organismal aging phenotypes is intricate and multifaceted. It remains unclear whether someone with longer telomeres can be considered biologically younger than someone half their age who possesses shorter telomeres. This observed variation in telomere length could potentially be attributed to natural differences in inherited telomere length and rates of telomere attrition, rather than serving as a definitive indicator of biological age. However, speculation that telomere shortening contributes to certain aging phenotypes has prompted the beauty industry to market anti-aging creams and serums claiming to elongate telomeres.
The telomeric DNA at chromosome ends is essential for preserving genomic stability – a task managed by the shelterin protein complex and by the enzyme telomerase. Telomerase, discovered in 1984 by Elizabeth Blackburn and Carol Greider, who were awarded the Nobel Prize, plays a pivotal role in maintaining and extending telomere length. During embryonic development, it ensures that telomeres in embryonic stem cells, germ cells, and other rapidly dividing cells do not shorten excessively. This activity is crucial for supporting the rapid cell divisions characteristic of this developmental phase. However, as development progresses towards birth and into postnatal life, telomerase activity is gradually silenced in most somatic (non-germline) cells and the telomeres gradually shorten with each cell division.
Triggering the DNA damage response when telomeres become critically short helps prevent the continued growth and spread of cancer cells
Cancer originates from mutations in a single cell that enable it to bypass the normal regulatory mechanisms of cell division. These mutations cause the cell to divide incessantly, producing more abnormal daughter cells and leading to the formation of a tumour. Therefore, the pathway involving telomere attrition and the subsequent activation of DNA damage responses seems to display all the characteristics of a tumour suppressor mechanism. By triggering the DNA damage response and inducing senescence or apoptosis when telomeres become critically short, this process helps prevent the continued growth and spread of cancer cells throughout the body.
This mechanism has been investigated by de Lange and other researchers. Recent findings from genome-wide association studies (GWAS) have identified genetic variants, primarily single nucleotide polymorphisms (SNPs), associated with longer telomere length in humans. These long telomere SNPs have been correlated with increased cancer risk, particularly for certain types of cancers. Normally, we are born with a certain telomere length that ensures the Hayflick limit activates at an appropriate time, minimising cancer risk by eliminating early cancer clones. However, being born with very long telomeres can delay the Hayflick limit, as it would require more divisions to shorten the telomeres. This delay extends the period during which oncogenic mutations can accumulate, thereby increasing the likelihood of forming immortalised cancer cells before the telomere-based tumour suppressor mechanism can eliminate them. Research results support such a hypothesis, with studies showing associations between genetic determinants of long telomeres and increased cancer risk.
Shelterin not only regulates the length of telomeres but also controls telomerase activity. When telomeres are long, shelterin assembles proteins like TRF1, TRF2, and POT1 on the telomeric DNA, which suppress telomerase, preventing it from extending telomeres further. This system maintains the normal telomere length seen at birth. If any part of this system is deficient, it can lead to longer telomeres and an increased cancer risk. Researchers at the de Lange lab at Rockefeller University in New York, and Radboud University Medical Center in Nijmegen, the Netherlands, studied cancer-prone families with mutations in the TINF2 gene, which produces a faulty TIN2 protein that cannot properly bind to telomeres. Using CRISPR to replicate these mutations, they found that the resulting cells had very long telomeres. Patient cell lines with these mutations also showed extremely long telomeres, much longer than usual. This telomere lengthening was seen in both cancerous and normal cells, suggesting that TINF2 mutations disrupt the telomere length control system, leading to longer telomeres at birth or in somatic cells.
Studies show associations between genetic determinants of long telomeres and increased cancer risk
Similar findings were observed with POT1 mutations, initially identified in familial melanoma cases, and studied by Dirk Hockemeyer at the University of California at Berkeley. Families with TINF2 or POT1 mutations often have a variety of cancers, such as colon, breast, thyroid, melanoma, chronic lymphocytic leukaemia, Hodgkin’s lymphoma, renal cell carcinoma, and glioma. Individuals with these mutations also tend to develop multiple primary cancers during their lifetime.
Bypassing the telomere protection
Although the tumour suppressor pathway imposed by telomere shortening is very powerful, it can nevertheless be bypassed, potentially resulting in cancer. One way this failure occurs is through the loss of p53, a crucial protein in the DNA damage response that leads to apoptosis and senescence. When cells lack p53, they ignore the senescence signals and continue to divide. Without telomerase, these cells can reach a state where many telomeres become too short to protect the chromosome ends. This status is known as telomere crisis. In the late 1990s, research by Ronald DePinho’s lab at MD Anderson Cancer Center in Houston, Texas, showed that telomere crisis in mice could cause chromosome abnormalities, such as translocations and losses. In the de Lange group, Teresa Davoli explored what happens to cells with permanent telomere damage. She deleted certain genes in mouse cells to disable their telomeres, and found that these cells completed one normal cell cycle but then re-entered the cycle incorrectly, leading to abnormal duplication and tetraploidisation (creating cells with four sets of homologous chromosomes instead of the usual two). Davoli observed the same effects in human mammary cells that lack p53, confirming the phenomenon across different cell types.
During telomere crisis, the formation of dicentric chromosomes is a crucial event. Instead of the usual single centromere – the point of constriction, which plays a key role in cell division – these chromosomes have two centromeres, which makes them unstable, leading to cycles of breakage and fusion that cause significant genetic instability. In the de Lange lab, John Maciejowski studied what happens to dicentric chromosomes formed from fused telomeres. He found that, during cell division, these chromosomes create a bridge that lasts for about 10 hours as the cells try to separate. Eventually, the bridge breaks due to loss of nuclear envelope integrity, allowing a DNA-cutting enzyme called TREX1 to enter and fragment the DNA. This fragmentation leads to a phenomenon called chromothripsis, where the genetic material is rearranged catastrophically.
Collaborating with Peter Campbell, the de Lange group sequenced cells that experienced telomere crisis and found that about half showed chromothripsis. By studying cells lacking TREX1, they discovered that this enzyme is crucial for this process. Without TREX1, the bridges took longer to resolve, and chromothripsis did not occur. Additionally, they found a specific mutation pattern, called kataegis, at the breakpoints of chromothripsis, which is caused by another enzyme called APOBEC3. Knocking out APOBEC3 prevented kataegis, confirming its role in this process.
During telomere crisis, most cells die due to the severe genomic instability, but a small fraction can survive by reactivating or upregulating telomerase. Telomerase can add telomeric repeats to eroded chromosome ends, stabilizing the genome. While telomerase activation is necessary for cells to escape telomere crisis, de Lange’s group has been exploring whether telomerase can also act at double-stranded breaks within the genome – a line of enquiry suggested by Charles Kinzig. Kinzig developed a method to study how telomerase affects DNA breaks. He found that telomerase can add telomeric repeats to these breaks, creating new structures called neotelomeres. When telomerase is too active, neotelomere formation increases, potentially aiding cancer cells by preventing harmful genetic instability. Understanding how neotelomeres form and function sheds light on how cancer cells cope with genetic chaos and offers insights into potential therapeutic strategies.
During the speech for her award acceptance talk in Trento, Italy Titia de Lange reminded us that if those skin rejuvenating creams really did elongate telomerase, they would be potentially carcinogenic. Fortunately, it is very unlikely that a cream could exert any effect on telomere length, which means we can safely keep on using them, if we like their smell.
About the Pezcoller–AACR 2024 award
Established in 1997, The Pezcoller Foundation–AACR International Award for Extraordinary Achievement in Cancer Research recognises scientists who have made major contributions to biological or translational cancer research.
The 2024 award was presented to Titia de Lange for discovering the molecular mechanisms by which telomeres protect chromosome ends, characterising the function of the shelterin protein complex, and demonstrating how loss of telomere protection results in aberrant genomic integrity and tumorigenesis.
With the collaboration of Francesca Albini
Featured image: Titia de Lange presenting at the Pezcoller Foundation‐AACR International Award for Extraordinary Achievement in Cancer Research Ceremony 2024 in Trento, Italy.
On the stage: Titia de Lange, Rockefeller University, New York, Pezcoller Foundation President Enzo Galligioni, AACR Past President Philip Greenberg, AACR CEO Margaret Foti, and Pezcoller Foundation Honorary President Gios Bernardi. Chromosome and telomeres are depicted on the screen.









