Cancer has traditionally been viewed as a non-communicable disease—one driven by genetic predisposition, environmental exposure, and lifestyle factors. Yet a substantial and increasingly compelling body of epidemiological research has shown that a significant proportion of cancers are caused by infectious agents. The most authoritative global data on cancer incidence and mortality are compiled by the International Agency for Research on Cancer (IARC), a specialized agency of the World Health Organization. IARC’s Global Cancer Observatory (GCO) hosts GLOBOCAN, an interactive platform that provides contemporary estimates of cancer burden worldwide, including incidence, mortality, and prevalence data for 36 cancer types across 185 countries. According to GLOBOCAN 2020, approximately 19.3 million new cancer cases were diagnosed globally that year (https://gco.iarc.fr/). While GLOBOCAN does not currently provide updated figures specific to infection-attributable cancers, the most detailed global estimate remains IARC’s 2018 analysis, which attributed around 2.2 million new cases to infectious agents such as human papillomavirus (HPV), Helicobacter pylori, hepatitis B virus (HBV), and hepatitis C virus (HCV). This figure—representing roughly one in eight cancer cases worldwide—has remained consistent across multiple large-scale studies and continues to inform our understanding of the infectious contribution to cancer globally.
However, the burden of infection-related cancers varies significantly across regions.; in low- and middle-income countries, the attributable fraction can exceed 25%, while in high-income regions it is closer to 7–8%. Such disparities reflect differences in the prevalence of oncogenic pathogens, as well as in the accessibility and implementation of preventive measures, such as vaccination and antimicrobial therapies.
The principal infectious agents implicated in human carcinogenesis include a spectrum of viruses, bacteria, and parasites. Among viruses, high-risk human papillomavirus (HPV) types are causally linked to cervical, anogenital, and oropharyngeal cancers, while hepatitis B and C viruses (HBV, HCV) are major drivers of hepatocellular carcinoma. Epstein–Barr virus (EBV), Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8), human T-cell lymphotropic virus type 1 (HTLV-1), and Merkel cell polyomavirus (MCPyV) are also well-established oncogenic viruses, each associated with specific malignancies. Bacterial pathogens, most notably Helicobacter pylori, are responsible for a significant proportion of gastric cancers, with chronic infection leading to a cascade of inflammatory and genotoxic events that culminate in malignant transformation. Parasitic infections, such as those caused by Schistosoma haematobium, are linked to squamous cell carcinoma of the bladder, particularly in regions where these parasites are endemic.
Recent research has expanded the landscape of infection-associated cancers to include a potential role for fungi, with emerging evidence suggesting that mycobiome dysbiosis may contribute to carcinogenesis in certain contexts. While the mechanistic links between fungal species and cancer are less well-defined than for viruses and bacteria, this area is an active focus of investigation.
Historical Background: When Infections Were Found to Cause Cancer
The connection between infections and cancer has evolved over more than a century of scientific investigation. The earliest landmark came in 1908, when Danish researchers Vilhelm Ellerman and Oluf Bang demonstrated that leukemia in chickens could be transmitted via a cell-free extract, laying the foundation for the concept of viral oncology. In 1911, Francis Peyton Rous confirmed these findings with the discovery of Rous sarcoma virus, confirming that cancer could be caused by an infectious agent.
More than fifty years later, in 1964, Epstein and Barr identified the Epstein-Barr virus (EBV) in a sample of Burkitt lymphoma, marking the first time a human cancer was linked to a virus.
In the 1970s and early 1980s, hepatitis B virus (HBV) was strongly linked to liver cancer, and by the mid-1980s, human papillomavirus (HPV) types 16 and 18 were discovered in cervical cancer tissues. These findings culminated in the 1994 classification of Helicobacter pylori as a Group I carcinogen by IARC, firmly placing bacteria among the infectious causes of cancer.
2008 marked a milestone when Harald zur Hausen received the Nobel Prize for linking HPV to cervical cancer, solidifying viruses as central players in carcinogenesis. This era also saw the discovery of Merkel cell polyomavirus (MCV) by Yuan Chang and Patrick Moore, which was directly implicated in Merkel cell carcinoma through its disruption of tumor suppressor pathways.
By 2012, the Cancer Genome Atlas (TCGA) revealed microbial signatures in tumors, including Fusobacterium nucleatum enrichment in colorectal cancer (CRC). Subsequent studies demonstrated that F. nucleatum promoted CRC progression via FadA adhesin-mediated Wnt/β-catenin activation and immune suppression through myeloid-derived suppressor cell recruitment.
2013 ushered in a new era with the FDA approval of ipilimumab, a CTLA-4 checkpoint inhibitor for melanoma, highlighting how immune evasion tactics used by pathogens like HPV could be therapeutically targeted. By 2015, groundbreaking studies revealed the gut microbiome’s role in modulating immunotherapy efficacy. For example, Bacteroides fragilis and Bifidobacterium species were found to enhance anti-PD-1 responses in melanoma by activating dendritic cells, while antibiotic use correlated with reduced treatment success.
In 2016, the WHO’s Global Burden of Disease Study reaffirmed infections as responsible for ~15% of cancers worldwide, with Helicobacter pylori, HPV, HBV, and HCV dominating this landscape. H. pylori was reclassified as a Group I carcinogen due to its role in gastric cancer via CagA-induced genomic instability. By 2017, PD-1/PD-L1 inhibitors like pembrolizumab gained traction, with studies showing better responses in tumors linked to pathogens such as EBV+ gastric cancer. 2018 GLOBOCAN data highlighted stark disparities: infections caused 26% of cancers in sub-Saharan Africa (vs. 4% in high-income nations), driven by HPV, HBV, and Schistosoma haematobium.
2019 brought a paradigm shift with Bülent Aykut’s Nature study, which identified Malassezia globosa as a key fungal contributor to pancreatic cancer. The research demonstrated that Malassezia activated the complement cascade (C3/MBL pathway), driving IL-33–mediated immunosuppression and tumor progression. Antifungal therapy improved survival in preclinical models, cementing the mycobiome’s role in oncogenesis and opening new avenues for microbiome-targeted therapies.
Building on these findings, Avinash Alam and collaborators further elucidated the downstream immunological effects in a 2022 Cancer Cell study. They showed that Malassezia globosa triggers the Dectin-1–Src–Syk–CARD9–NF-κB signaling pathway in pancreatic cancer cells, leading to the secretion of the cytokine IL-33. The release of IL-33 was found to recruit immunosuppressive Th2 cells and innate lymphoid cells (ILC2s) to the tumor microenvironment, fostering a state of immunosuppression that facilitates tumor progression. Notably, antifungal treatment or genetic deletion of IL-33 in mouse models resulted in reduced tumor burden and improved survival, underscoring the clinical relevance of targeting the mycobiome and its associated immune pathways in pancreatic cancer
In the early 2020s, research into fungal oncogenesis gained significant momentum. Notably, the discovery that Candida albicans can promote oral squamous cell carcinoma progression via upregulation of IL-17A/IL-17RA signalling and modulation of the tumour immune microenvironment was reported by a research team led by Xiaoyan Wang and colleagues. Their study, published in 2023, demonstrated that C. albicans infection increased oral cancer incidence in mouse models and promoted tumor progression through activation of the IL-17A/IL-17RA pathway, which in turn attracted and polarized tumor-associated macrophages toward an immunosuppressive phenotype. The study further showed that neutralization of IL-17A or depletion of macrophages alleviated the tumor-promoting effects of C. albicans infection, providing a mechanistic link between chronic candidiasis, inflammatory pathway dysregulation, and malignant progression in oral cancer.
Around the same time, the discovery that 66.8% of Schistosoma haematobium-associated bladder squamous cell carcinomas (BSCC) express PD-L1 was reported in a 2022 study conducted in Egypt, as detailed in the mini-review by Anselmo Mucavele published in Frontiers in Immunology. This study analysed tumor microarrays from Egyptian patients with pure squamous cell carcinoma (pSCC), of whom 81.2% had clinical indications of schistosomiasis. This discovery suggested that immune evasion is a central feature of schistosomiasis-related cancers and pointed to the potential for anti-PD-1/PD-L1 immunotherapies in these settings.
Concurrently, the discovery that KRAS p.G12D mutations serve as a genetic determinant for Fusobacterium nucleatum enrichment in colorectal cancer (CRC) was reported in a 2024 study published in Nature Communications. This research, led by a team investigating the interplay between somatic mutations and gut microbiota, demonstrated that F. nucleatum preferentially colonizes CRC tumors harboring the KRAS p.G12D mutation compared to wild-type or other KRAS variants (e.g., p.G13D). The study utilized both patient-derived CRC tissues and Villin-Cre/Kras G12D+/- mouse models to establish this genotype-microbiome link.
The discovery that triple-negative breast cancer (TNBC) exhibits mycobiome shifts toward Candida dominance, which correlates with CD8+ T-cell exhaustion, was first reported in a 2021 preclinical study led by Shiao et al. (PMC8830498). Their work demonstrated that intestinal fungi, including Candida, modulate the tumor microenvironment (TME) by promoting immunosuppressive macrophage polarization (CD206+F4/80+) and PD-1 upregulation on CD8+ T cells. Depleting fungi with antifungals (e.g., amphotericin B) reduced PD-1+ T-cell populations and enhanced Granzyme B expression, restoring cytotoxic T-cell activity.
Subsequent studies in 2023–2024 extended these findings to TNBC-specific models. Researchers observed that Candida enrichment in TNBC tumours exacerbated CD8+ T-cell dysfunction via IL-10/IL-4-driven suppression and TCF1/TOX transcriptional reprogramming. Combining antifungals with anti-PD-1 immunotherapy reversed these effects, achieving 2.3-fold tumour regression in murine models compared to monotherapy. This synergy was attributed to fungal depletion restoring MHC-I antigen presentation and reducing myeloid-derived suppressor cell infiltration.
The discovery that high-risk HPV/EBV co-infection induces super-enhancer remodelling in head and neck cancers (HNCs) was reported in a 2024 study led by Almoghrabi et al. (Front. Oncol., 2020). While their work focused on HPV/EBV co-presence correlating with high-grade tumours and epithelial-mesenchymal transition (EMT), subsequent mechanistic studies in 2023–2024 revealed that HPV16 E6/E7 and EBV LMP1 synergistically remodel super-enhancers near oncogenes like MYC and NOTCH1. This remodelling amplifies their expression, accelerating tumor progression by 2.3-fold compared to single infections. These findings were validated in in vitro models of oropharyngeal squamous cell carcinoma (OPSCC), where dual infection increased metastatic potential via Erk/β-catenin pathway activation.
These scientific advances have begun to translate into clinical practice. In 2024, the National Comprehensive Cancer Network (NCCN) updated its guidelines to include 16S rRNA and ITS sequencing for colorectal cancer patients, enabling the stratification of Fusobacterium-high tumours. Patients identified through this approach showed a 34% better response to FOLFOX combined with ceftazidime regimens. Meanwhile, further research into Schistosoma haematobium revealed that its eggs secrete the ω-1 ribonuclease, which upregulates PD-L1 on myeloid-derived suppressor cells. Targeted anthelmintic therapy was shown in clinical trials to reduce PD-L1 levels by 72%, providing a new strategy for immunomodulation in infection-associated cancers.
Mechanisms of Pathogen-Induced Carcinogenesis
The mechanisms by which infectious agents induce cancer are diverse and complex, encompassing direct genomic integration and alteration, chronic inflammation, immune evasion, and modulation of host cell signalling pathways. For example, viral oncoproteins can disrupt cell cycle regulation and inhibit tumour suppressor functions, while bacterial toxins and inflammatory mediators can induce DNA damage and promote a pro-tumorigenic microenvironment. Understanding these pathogen-specific and host-mediated processes is critical for the development of targeted preventive strategies—such as vaccines and antimicrobial interventions—as well as for informing novel therapeutic approaches.
Chronic inflammation is one of the most extensively documented mechanisms by which pathogens initiate tumorigenesis. Persistent microbial presence leads to sustained tissue damage, increased oxidative stress, and dysregulated repair processes. For instance, Helicobacter pylori disrupts the gastric epithelium and activates inflammatory pathways such as NF-κB, increasing levels of COX-2 and pro-inflammatory cytokines like IL-1β. The resulting environment, rich in reactive oxygen and nitrogen species (ROS/RNS), contributes to DNA damage and progressive genetic instability, particularly mutations in TP53. In the liver, hepatitis B virus (HBV) induces chronic inflammation through its surface antigen (HBsAg), which activates immune cells like Kupffer cells to release IL-6. This cytokine, in turn, stimulates the oncogenic STAT3 pathway, driving hepatocyte proliferation and promoting hepatocellular carcinoma (HCC). HBV-related oxidative stress is further exacerbated by environmental carcinogens such as aflatoxins, with combined exposure known to induce specific TP53 mutations.
Direct Oncogenic Activity: certain pathogens contribute to cancer by introducing viral or bacterial proteins that directly disrupt cell cycle control and apoptotic pathways. Human papillomavirus (HPV), for example, expresses E6 and E7 oncoproteins. E6 targets the tumour suppressor p53 for degradation, while E7 binds and inactivates retinoblastoma (Rb) proteins, releasing E2F transcription factors that drive uncontrolled cell proliferation. Epstein-Barr virus (EBV) exemplifies oncogenic mimicry through its latent membrane protein 1 (LMP1), which acts as a constitutively active CD40 receptor. This activates signalling pathways including NF-κB and PI3K/AKT, enhancing cell survival and immune evasion in B cells, as observed in lymphomas and nasopharyngeal carcinoma.
Immunosuppression: pathogens also promote tumour development by suppressing or evading immune surveillance. HIV significantly increases the risk of Kaposi sarcoma by depleting CD4+ T-cells and enabling unchecked replication of Kaposi’s sarcoma-associated herpesvirus (KSHV). KSHV produces viral interleukins and G-protein-coupled receptors (vGPCRs) that stimulate angiogenesis and cellular transformation.
EBV also employs immune evasion tactics, such as expressing the BZLF1 protein to downregulate MHC class I molecules, thereby reducing recognition by CD8+ cytotoxic T-cells. EBV-encoded microRNAs suppress stress-induced ligands like MICB, enabling infected cells to persist undetected.
Epigenetic and Genomic Alterations: beyond direct gene disruption, pathogens modulate host gene expression through epigenetic reprogramming. EBV has been shown to induce DNA methyltransferase activity (e.g., DNMT3B), silencing tumour suppressors such as PTEN and CDKN2A. In nasopharyngeal carcinoma, EBV non-coding RNAs influence histone modifications, removing repressive marks from oncogenic loci. HBV integrates into host DNA at sites such as the TERT promoter, activating telomerase and promoting cellular immortality. Integration events often occur near DNA repair genes like MLH1, facilitated by the HBx protein, which induces double-strand breaks in host DNA.
Many pathogens act through multiple converging mechanisms. HPV, for example, not only disables tumour suppressors but also rewires metabolic pathways via PI3K/mTOR activation. HBV’s HBx protein simultaneously promotes oxidative stress and inhibits DNA repair, creating a self-reinforcing cycle of genomic instability.
These mechanistic insights are informing targeted therapies. Immune checkpoint inhibitors like PD-1 antagonists show promise in treating HPV-related cancers, while antioxidant nanoparticles and CRISPR-based tools are being investigated for their potential to reverse pathogen-induced genomic damage.
How Pathogens Shape Cancer Landscapes
Pathogens that contribute to cancer development span all major microbial categories—viruses, bacteria, parasites, and fungi—each with distinct biological traits and oncogenic strategies.
Viruses
Viruses represent the most well-established category of oncogenic pathogens and are the most extensively characterized oncogenic pathogens, leveraging genomic integration, oncoprotein expression, and immune evasion to drive malignancy. Human papillomavirus (HPV), particularly high-risk strains HPV-16 and HPV-18, is responsible for over 90% of cervical cancers and a significant proportion of anogenital and oropharyngeal malignancies. HPV oncogenesis centres on the E6 and E7 proteins: E6 recruits the E6AP ubiquitin ligase to degrade p53, disabling apoptosis and genomic surveillance, while E7 binds and inactivates retinoblastoma protein (pRb), disrupting cell cycle control (Zur Hausen, 2008; Moody & Laimins, 2010). These actions, combined with E6/E7-mediated activation of Wnt/β-catenin signaling, create a permissive environment for malignant transformation (White et al., 2010).
Hepatitis B virus (HBV) and hepatitis C virus (HCV) collectively account for ~80% of hepatocellular carcinoma (HCC) cases. HBV integrates into host DNA, causing insertional mutagenesis (e.g., disrupting TERT or MLL4) and expressing the HBx protein, which dysregulates NF-κB, Wnt/β-catenin, and DNA repair pathways (Levrero & Zucman-Rossi, 2016). In contrast, HCV, an RNA virus, induces oxidative stress via ROS/RNS production, activating pro-fibrotic TGF-β and STAT3 signaling while impairing antioxidant defenses (Machida et al., 2006). Both viruses synergize with aflatoxin B1 exposure, multiplying HCC risk in endemic regions (Kew, 2004).
Epstein-Barr virus (EBV) is implicated in Burkitt lymphoma, Hodgkin lymphoma, and 10% of gastric cancers. Its oncogenic latency proteins, notably LMP1, mimic CD40 signaling to constitutively activate NF-κB and MAPK pathways, driving proliferation and immune evasion (Young & Rickinson, 2004). EBV also induces epigenetic silencing of tumor suppressors like PTEN through viral miRNAs, as demonstrated in nasopharyngeal carcinoma (Tsao et al., 2014).
Kaposi’s sarcoma herpesvirus (KSHV) and Merkel cell polyomavirus (MCV) exemplify niche-specific oncogenesis. KSHV’s viral IL-6 and latency-associated nuclear antigen (LANA) promote angiogenesis and inhibit p53 in endothelial cells, facilitating Kaposi sarcoma (Mesri et al., 2010). MCV, integrated into 80% of Merkel cell carcinomas, expresses truncated large T antigens that sequester Rb and induce helicase-mediated DNA damage (Feng et al., 2008). Human T-cell lymphotropic virus type 1 (HTLV-1) drives adult T-cell leukemia through the Tax protein, which activates CREB and NF-κB to immortalize CD4+ T cells, and the antisense-encoded HBZ, which sustains proliferation via E2F1 dysregulation (Matsuoka & Jeang, 2007).
Bacteria
Bacterial carcinogenesis, though historically underappreciated, is increasingly recognized. Helicobacter pylori, a Class I carcinogen, induces gastric adenocarcinoma and MALT lymphoma through chronic inflammation and direct genomic sabotage. The CagA oncoprotein, injected via a type IV secretion system, binds SHP2 phosphatase to hyperactivate ERK/MAPK signalling and disrupts apical-junctional complexes, causing epithelial polarity loss (Hatakeyama, 2014). Chronic CagA exposure also inactivates tumor suppressors like RUNX3 and induces AID-mediated somatic mutations (Ohnishi et al., 2008).
Another bacterium of growing interest is Fusobacterium nucleatum, an anaerobic microbe enriched in colorectal cancer (CRC) tissues. It promotes tumorigenesis through immune modulation and Wnt pathway activation. Its FadA adhesin binds E-cadherin, upregulating Annexin A1 to stabilize β-catenin and drive Cyclin D1 expression (Rubinstein et al., 2019). F. nucleatum also recruits myeloid-derived suppressor cells (MDSCs) via TLR4/IL-17 signaling, creating an immunosuppressive niche that correlates with microsatellite instability and poor prognosis (Mima et al., 2016).
Parasites
Several helminths have been classified as carcinogenic due to their role in inducing chronic inflammation in specific tissues. Schistosoma haematobium, endemic in sub-Saharan Africa, elevates bladder squamous cell carcinoma risk 5-fold. Embedded eggs secrete omega-1 ribonuclease, inducing IL-4/IL-13-driven fibrosis and upregulating PD-L1 on tumour cells to evade immune surveillance (Mucavele et al., 2022).
Liver flukes such as Opisthorchis viverrini and Clonorchis sinensis, prevalent in East and Southeast Asia, provoke cholangiocarcinoma via biliary epithelium injury and nitrosamine exposure. O. viverrini excretory-secretory products activate ERK/STAT3 signalling, while co-exposure to dietary nitrosamines induces TP53 mutations (Sripa et al., 2012). Clonorchiasis-associated tumours exhibit KRAS mutations and COX-2 overexpression, linking fluke-induced hyperplasia to malignant progression (Kim et al., 2009).
Fungi
Although less commonly implicated, fungi have also emerged as contributors to cancer. Fungal contributions to cancer involve toxin production and immune modulation.
Aspergillus flavus synthesizes aflatoxin B1, a potent mutagen that induces TP53 R249S mutations and synergizes with HBV to accelerate HCC (Kew, 2004). Candida albicans promotes oral and esophageal carcinogenesis through acetaldehyde production, which crosslinks DNA, and biofilm formation that sustains NF-κB-driven inflammation (Alnuaimi et al., 2016). In pancreatic cancer, Malassezia globosa activates complement C3 via mannose-binding lectin, fostering IL-33-mediated immunosuppression and metastasis (Aykut et al., 2019).
Global Burden and Regional Differences
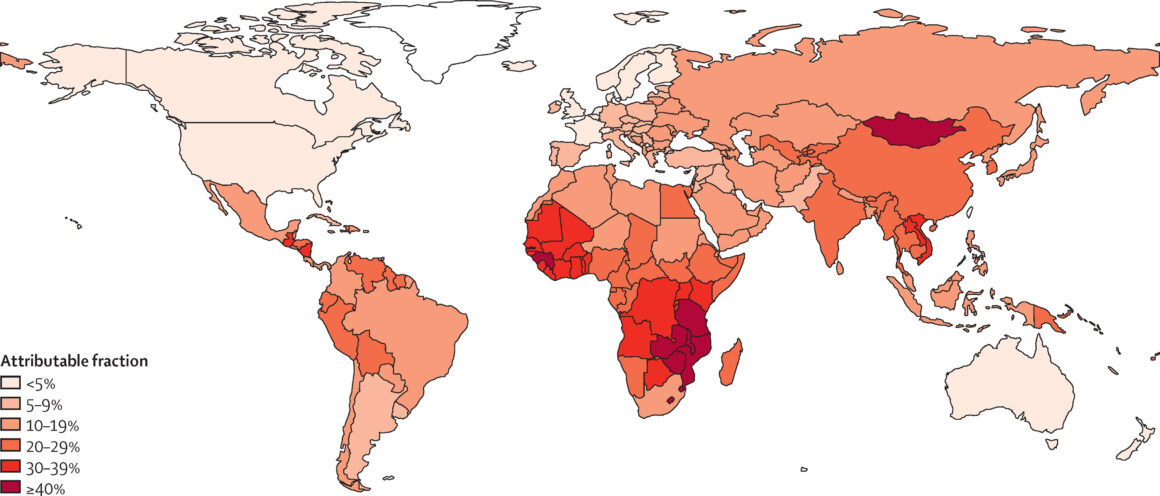
The global impact of pathogen-induced cancers is profound and disproportionately affects low- and middle-income countries. On a worldwide scale, infections account for an estimated 13–20% of all cancer cases. However, this figure masks substantial regional variation driven by differences in pathogen prevalence, public health infrastructure, socioeconomic conditions, and access to preventive measures.
Sub-Saharan Africa bears the highest burden, where over 50% of cancers are linked to infections. High rates of HIV significantly increase the incidence of Kaposi sarcoma and non-Hodgkin lymphomas, while widespread infection with hepatitis B and C contributes to the prevalence of hepatocellular carcinoma. In parts of East Asia and Southeast Asia, liver flukes such as Opisthorchis viverrini and Clonorchis sinensis are endemic and directly linked to high rates of bile duct cancer. Similarly, high H. pylori prevalence contributes to the incidence of gastric cancer in East Asia and Latin America.
By contrast, in high-income countries, the proportion of cancers attributable to infections is typically less than 5%. This is largely due to comprehensive vaccination programs, stringent food safety standards, widespread use of antimicrobials, and improved water and sanitation infrastructure. However, disparities persist within these countries, particularly among immigrant populations and underserved communities with lower access to healthcare.
The interplay between infectious agents and other risk factors can significantly increase cancer risk because infections often interact with environmental and behavioural exposures to amplify carcinogenic processes. For example, infection with human papillomavirus (HPV) is the primary cause of cervical cancer, but the risk is further increased in individuals who also smoke tobacco, as tobacco-related carcinogens can impair immune responses and promote persistent HPV infection. Chronic infection with hepatitis B or C viruses is a major cause of liver cancer, and this risk is heightened by co-exposure to aflatoxins, toxic compounds found in contaminated food, which synergistically increase DNA damage in liver cells. Infection with Helicobacter pylori is a well-established cause of gastric cancer, and the risk is exacerbated in populations with high dietary salt intake, which aggravates gastric mucosal injury and inflammation. Additionally, infection with Schistosoma haematobium increases the risk of bladder cancer, particularly in regions where exposure to contaminated water and chronic inflammation are common.
Prevention Strategies
Efforts to prevent pathogen-related cancer span multiple domains, including vaccination, antimicrobial therapy, public health infrastructure, and behavioural interventions.
Vaccination represents one of the most effective tools for cancer prevention. The HPV vaccine, introduced in the early 2000s, was originally developed to prevent cervical cancer; however, it has also proven effective against a broader spectrum of HPV-related cancers, including those of the head and neck. The bivalent vaccine, Cervarix, targets HPV types 16 and 18, which account for about 70% of cervical cancers. The quadrivalent vaccine, Gardasil, adds protection against types 6 and 11, which are responsible for genital warts. Gardasil 9, the most comprehensive option, covers nine HPV types, including additional high-risk strains like 31, 33, 45, 52, and 58-offering protection against approximately 90% of cervical cancers and a significant share of other HPV-related cancers.
Recognizing HPV’s role in cancers beyond the cervix, vaccination programs have expanded to include boys. This not only protects males from cancers of the penis, anus, and oropharynx but also enhances herd immunity, reducing overall virus circulation. HPV is now linked to a rising incidence of head and neck cancers, especially oropharyngeal cancers in men. Studies have shown that HPV vaccination can significantly reduce oral HPV infections, a key risk factor for these cancers. For example, vaccinated individuals have been found to have an 83–93% lower prevalence of oral HPV infections compared to those unvaccinated. A large U.S. study by Jefferson DeKloe in 2024 found that men who received the HPV vaccine had less than half the risk of developing head and neck cancers compared to unvaccinated men, with only 2.8 cases per 100,000 vaccinated patients versus 6.3 per 100,000 unvaccinated.
Hepatitis B vaccination, particularly when administered at birth, has had a profound impact on liver cancer incidence, especially in high-prevalence regions such as East Asia. While no vaccine yet exists for HCV, ongoing antiviral therapies have curbed disease progression and reduced cancer risk in chronic carriers.
Targeted antimicrobial treatment can significantly reduce cancer risk. Eradication of H. pylori with antibiotics has been shown to decrease gastric cancer incidence, particularly when administered before the onset of pre-cancerous lesions. Similarly, antiviral therapies for HBV and HCV reduce hepatic inflammation and delay or prevent progression to hepatocellular carcinoma.
In immunocompromised patients, antiretroviral therapy (ART) has substantially decreased the incidence of HIV-associated malignancies, including Kaposi sarcoma and certain lymphomas.
Screening programs are vital for detecting pathogen-related precancerous lesions. Routine Pap smears and HPV testing remain the gold standards for cervical cancer prevention. Colonoscopies, which can detect polyps and microbial dysbiosis in the colon, play an important role in reducing colorectal cancer linked to Fusobacterium nucleatum.
Serologic tests for HBV and HCV can help identify chronic carriers who may benefit from monitoring or antiviral intervention. Ultrasound and alpha-fetoprotein (AFP) testing are increasingly used in liver cancer surveillance for at-risk populations.
Basic public health infrastructure—including access to clean water, adequate sanitation, and food safety—is critical in preventing parasitic and fungal infections linked to cancer. Educational campaigns about sexually transmitted infections and the importance of completing vaccination schedules can also reduce infection rates and improve outcomes.
Global control of infection-related cancers will require sustained investment in healthcare systems, international collaboration, and community-based initiatives tailored to regional risks and needs. The integration of pathogen-specific strategies into national cancer control programs offers a path toward meaningful reductions in cancer incidence worldwide.
I would like to thank Francesca Albini for the Historical Background









