
RAS oncogenes and their proteins have central roles in almost all cancers, including leukaemias, multiple myelomas, skin cancers and many solid tumours, making the RAS protein family an ideal cancer target. But efforts to develop clinically efficacious drugs to target RAS, starting in the early 1990s, met with unforeseen hurdles and disappointing clinical progress. The scientific and clinical setbacks, however, unexpectedly revealed the importance of the immune system in unlocking potential synergies of combining RAS targeting with immunotherapy approaches, which were recently highlighted in Nature, and could prove important.
A key role in growth and proliferation
RAS proteins are important for normal cellular development because they are pivotal in driving the growth and proliferation of cells and, as recently reported, also normal cell migration. They act as binary switches cycling between the ‘off’ (inactive, GDP-bound) state and the ‘on’ (activated GTP-bound) state.
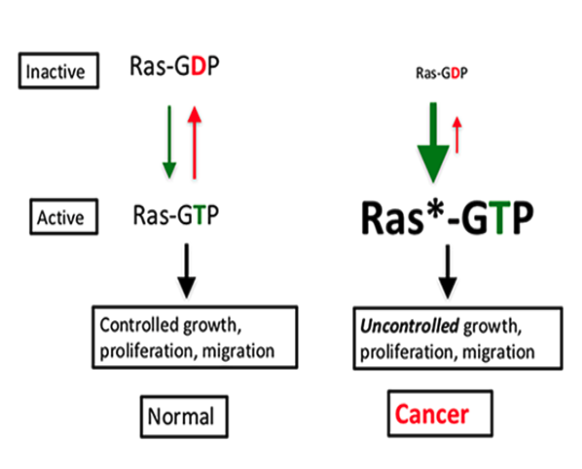
Credit: Jim Hartley, NCI RAS Initiative
RAS proteins are important in normal cell growth, proliferation and cell migration. When RAS binds guanine nucleotide exchange factors (GEFs), it is converted to the GTP-bound active or switched ‘on’ state (green down arrow). Conversely, the protein is rendered inactive by hydrolysis to RAS-GDP via GTPase activating proteins (GAFs) and turned ‘off’ (red up arrow). In cancer cells, the mutated RAS* is stuck in the active or ‘on’ state, driving unregulated cell growth and proliferation. RAS proteins lack intrinsic enzyme activity, are small in size and lack surface ‘pockets’ making them hard to target. Improved understanding of RAS signalling and regulation has now created opportunities to address this situation.
Active RAS is a potent signalling complex that stimulates many downstream effector molecules, such as the intracellular kinases PI3K and Raf, which are known to play a role in many cancers. In normal cellular conditions, the RAS protein is found in more limited amounts in its active state, presumably to balance signalling output. However, when RAS becomes mutated, the resulting protein gets stuck in the activated state, which drives uncontrolled cell growth that ultimately leads to cells becoming cancerous.
A ubiquitous oncogene
Evidence over several decades shows that between 20% and 30% of cancers are driven by mutations in the RAS gene. The main members of the RAS gene family — KRAS, HRAS, and NRAS — encode proteins that play a pivotal role in cytoplasmic cell signalling. Multiple tumour types, including lung, breast, ovarian, and colorectal, contain RAS mutations. The most prevalent RAS-oncogenic driven cancers are those of the lung, colon and pancreas (see figure below). Globally, it is estimated there are 10 million cancer deaths annually, and 10-15% of these deaths can be considered RAS-oncogene driven. Therefore, RAS became one of the prime targets for oncology research and drug targeting.
Lung cancer is the most prevalent of malignancies; while the decline of this cancer in the Western world is notable, it is still high. Specifically, smoking is mainly associated with high KRAS mutation rates in lung cancers. KRAS mutations are also common in pancreatic adenocarcinomas and colorectal cancers, while NRAS mutations are more prevalent in melanomas, thyroid cancers and leukaemias, and HRAS in kidney and thyroid tumours. Colon cancer is also becoming more common, but pancreatic cancers are among the hardest to treat, with fairly stagnant clinical progress.
RAS mutations are common across a wide range of cancers
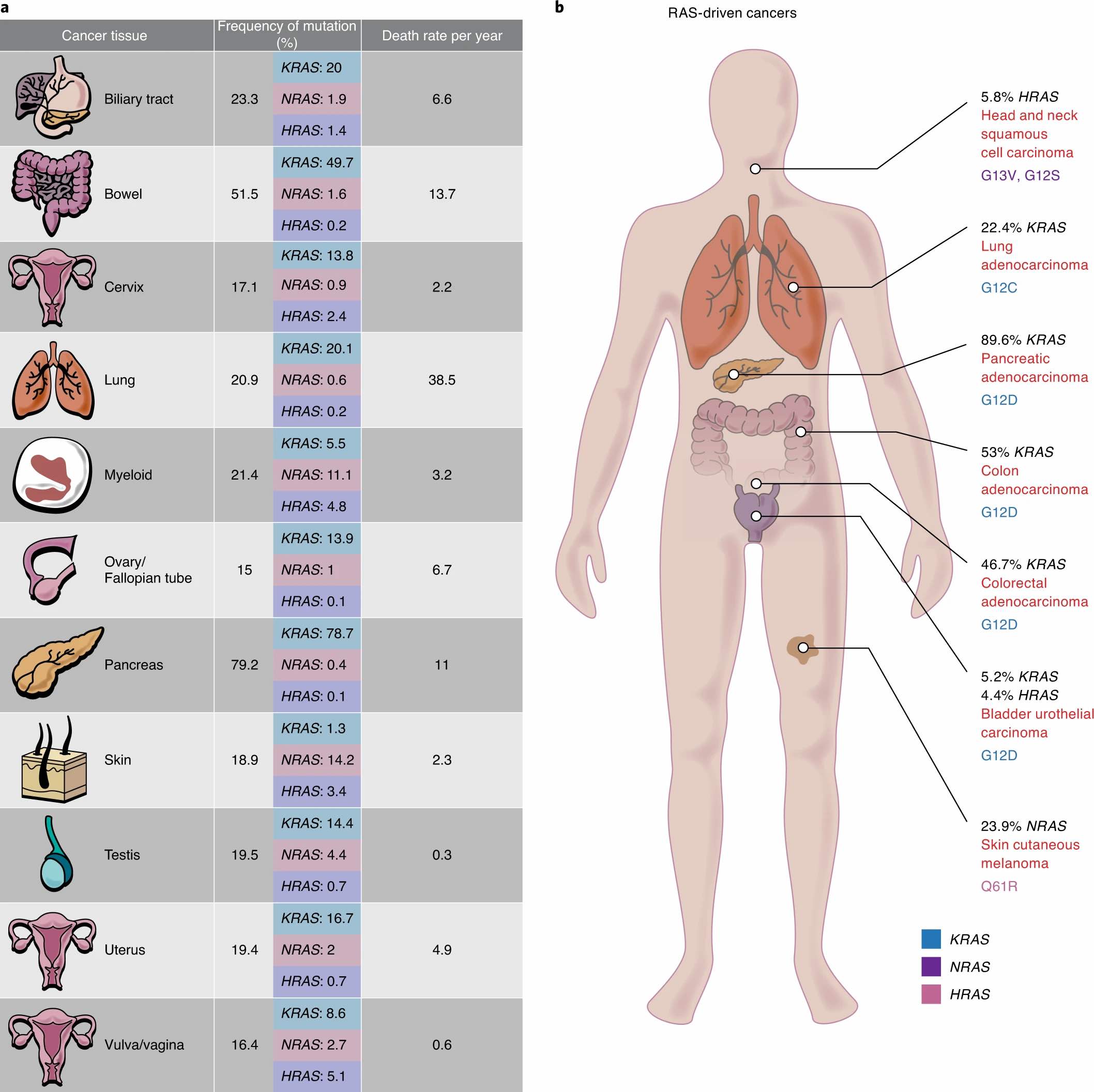
Source: Suman Mukhopadhyay et al, The metabolic landscape of RAS-driven cancers from biology to therapy, Figure 1 Mar 24, 2021 Springer Nature. Republished with permission. © Nature 2021
RAS – the undruggable target?
For decades, RAS was considered ‘undruggable’, chiefly because it was very difficult to identify a surface or ‘pocket’ that an inhibitor drug could bind to. RAS proteins are small (around 20kDa), and they lack intrinsic enzyme activity, which means they cannot be targeted using the same mechanisms that have been deployed with tyrosine kinase inhibitors.
Secondly, RAS has a fundamental role in cell growth, so while it made sense to target mutated RAS that was driving unrestrained proliferation, blocking wild-type RAS – as the first generation of RAS inhibitors did – turned out to be as toxic as chemotherapy, because they also targeted growing non-cancerous cells. What was needed was a drug that could selectively target mutated RAS.
Finding a drug that could target the altered protein turned out to be exceptionally tricky. It wasn’t until 2021 that the first such drug was approved by the US regulator, the FDA. The intervening decades saw a variety of alternative approaches to controlling cancers driven by RAS mutations, which involved modifying the protein by other mechanisms or indirect targeting of proteins downstream or growth factor receptors upstream of RAS.
Direst RAS modification: farnesyltransferase inhibitors
One initial approach that held high hopes was to prevent the farnesylation of RAS, which is required to turn the protein into its biologically active form. Farnesyltransferase inhibitors (FTIs) were quickly developed, and many showed promise in preclinical studies, suppressing tumour growth with little toxicity. But FTIs showed more subdued effects in the clinic.
It turned out that, while the FTIs hit their intended targets, the tumour cells signalled other pathways into action, rendering the blockade ineffective (often referred to as feedback loops). This was one of the early lessons of the limitations of even the best designed targeted drugs, due to cancer’s ability to develop resistance by using alternative ‘escape’ pathways.
When the first clinical trials of these agents flopped, enthusiasm for FTIs soon dwindled, although they may be staging a come-back with a new generation of FTIs, such as tipifarnib for treating head and neck cancer, which look more promising and have entered phase III testing.
Targeting downstream of RAS
Raf and MEK
Targeting signals through a network of effector pathways has been the subject of intense drug discovery. RAF, the downstream kinase of RAS, seemed a logical next target for treating KRAS-mutated cancers. Sorafenib, the first BRAF inhibitor to enter clinical trials, while effective for some cancers, showed limited benefit for patients with KRAS-mutated lung tumours. Second-generation inhibitors, such as vemurafenib, while showing efficacy in some melanomas that harboured a BRAF driver mutation, were also found to be ineffective in cancers with mutated RAS. Notably, studies showed survival rates were lower in patients treated with epidermal growth factor receptor (EGFR) inhibitors (see below), when BRAF or KRAS were mutated, indicating that these mutations are negative predictive markers to EGFR therapies. Presumably, the BRAF and KRAS mutations when used with receptor inhibitors paradoxically boost RAF signalling, presumably via alternative pathways.
Since resistance mechanisms via alternative signalling seemed to hinder progress in attempts to target KRAS, focus then shifted to targeting other proteins in the RAS signalling pathway. Chief among them was MEK (mitogen activated protein kinase kinase), which functions downstream of RAF. But while MEK inhibitors, such as trametinib, also found a role in treating BRAF mutated cancers, again they proved ineffective in the downstream blocking of RAS signalling. MEK inhibitors, it seems, reduce the feedback suppression of upstream signalling, which then leads to increased receptor tyrosine kinase signalling, thereby counteracting the inhibition of MEK.
In short, huge efforts made over several decades to finding ways to control RAS-driven cancers by hitting downstream targets have yielded limited success – although much has been learnt about the complexity of cancer in the process. Reasons that have been given to explain the disappointing results include: insufficient target inhibition, a lack of therapeutic window (too ineffective at tolerable doses, too toxic at effective doses), feedback or crosstalk of cellular responses, and tumour heterogeneity.
Targeting upstream of RAS: growth factor receptors
As with many anticancer approaches, the off-target effects and toxicities noted with the first-generation RAS inhibitors, as well as acquired resistance, prevented the clinical success of these agents. This resulted in a downturn in enthusiasm for targeting RAS, which coincided with an increased appetite for developing inhibitors to growth factor receptors, such as EGFR. However, with first-generation EGFR tyrosine kinase inhibitors (TKIs), considerable skin toxicity affected treatment compliance. Next-generation TKIs, such as osimertinib, which was efficacious in lung cancer patients with the T790M EGFR mutation, has shown promise in the clinic .
The EGFR inhibitor cetuximab was the first targeted drug to show meaningful benefits in patients with colorectal cancers and also in head and neck cancers; however, as noted earlier, only in those with wild-type KRAS. Non-responders were those with mutated G12C KRAS, highlighting how successful EGFR inhibition met with limited success in patients with mutant KRAS.
The G12C mutation is less common in colorectal cancer compared with lung cancer, which is why the promising data with EGFR inhibition seen in colorectal cancer did not translate to other cancer sites. Current options include combinations with BRAF inhibitors and immunotherapies, but resistance continues to compound such combinations longer term. Collectively, we have not figured out how to test the multitude of combinations and so far the results have been fairly unimpressive.
Hitting mutated RAS – the breakthrough
While all these efforts were going on to control RAS-driven cancers by hitting downstream or upstream of the mutated gene, a few laboratories stuck to the original goal of finding a way to directly target mutated RAS. The first breakthrough came in 2013, when Kevan Shokat, at UCSF, California, discovered a drug-like molecule that could bind to mutant G12C KRAS – the most common mutation in non-small-cell lung cancer. The G12C mutation refers to a mutation at codon 12 of the KRAS gene, where glycine is replace by the amino acid cysteine.
The drug works by irreversibly targeting the cysteine residue in G12C mutant KRAS in its inactive state, which has the effect of keeping the gene permanently locked in its ‘switched off’ state. This research advance prompted Amgen and Mirati Therapeutics to develop two direct KRAS-G12C inhibitors: sotorasib and adagrasib.
In 2021, the FDA granted accelerated approval of sotorasib for the treatment of patients with NSCLC. This was based on data from a phase II trial showing responses (tumour shrinkage) in 37% of patients, with an 11-month median gain in progression-free survival. An ongoing phase III trial of sotorasib versus docetaxel in G12C KRAS NSCLC, however, has so far proved disappointing, with no overall survival benefit observed at the 24-month time point assessment.
In December 2022, adagrasib received accelerated approval for treating locally advanced or metastatic NSCLC. This conditional approval followed results of the KRYSTAL-1 multicentre, single-arm, open-label trial, which showed an objective response rate of 43% and median progression-free survival of 6.5 months. More trials using KRAS-G12C inhibitor monotherapy, or combination approaches with immunotherapies, for the treatment of patients with NSCLC and other solid tumours are underway.
Sadly, as with most cancer therapies, multiple mechanisms of resistance to KRAS G12C inhibitors are already emerging, which means combination treatments will probably be needed for a lasting impact. Resistance raises several important questions about the clinical application of KRAS G12C inhibitors.
Worth the wait?
Julian Downward, Associate Research Director at the Francis Crick Institute, in London, has had an interest in RAS for more than 35 years, having done his post doctoral research at the Massachusetts Institute of Technology in 1986, with Robert Weinberg, who identified the RAS gene family as the first oncogenes in human cancers. Downward is delighted that decades of efforts to inhibit mutant KRAS have finally paid off. But he’s also distinctly cautious about what they may be able to deliver for patients and how.
In an interview with Cancerworld, he argued that there are too many iterations of the same KRAS-G12C inhibitor in development, and he stressed the significant obstacles to their efficacy posed by intratumoural heterogeneity and the numerous escape mechanisms of cancer cells. That said, research carried out in his and other labs at the Francis Crick Institute, has shown that, in mice models with RAS-driven lung cancers, RAS inhibition can result in significant and unexpected changes in the tumour microenvironment (TME), which could open new avenues. This is where Downward hopes some real progress could be made.
The curious story of RAS and tumour immune response
Over recent years, Downward’s lab has been developing novel mouse models for RAS-driven lung cancers, in which, in contrast to traditional models, the mice retain a fully functioning immune system. This opened the way to evaluate how RAS inhibition interacts with the immune system and the potential of combined RAS inhibitor–immunotherapy combinations.
It turns out that, in addition to driving uncontrolled growth, the RAS oncoprotein also affects the tumour microenvironment (TME). RAS mutations cause an immunosuppresive TME, and so inhibiting RAS tumours showed profound changes in all TME cells, such as the stroma, as well as accompanying cytokine and chemokine transcriptional expression that manipulates the local microenvironment.
Inhibition of RAS collapses the protection of tumours from the immune system, and it could be this effect, rather than the original goal of blocking its signal for cell proliferation, that could offer the most significant therapeutic impact from inhibiting mutated RAS. Combining RAS inhibitors with immune modifiers is a hugely active area of ongoing clinical research that might finally open a new era for cancer therapy.
An ancient retrovirus to the rescue
Studies in both mouse models and human tissue samples of KRAS-driven lung cancer revealed a most unexpected mechanism mediating the link between RAS inhibition and changes in the immune TME. The heightened immune response was led not by T lymphocytes – which directly kill cells that have already been infected by a foreign invader – but by B lymphocytes – which produce antibodies to fight bacteria and viruses. It turns out that the signal summoning B cells to the TME is a protein coming from endogenous retroviruses – these are integrated remnants of viral DNA that, in the process of evolution, have been incorporated into normal human cells, and form about 5% of the human genome. Inhibiting mutated RAS seems to reactivate these dormant viruses.
A strong presence of B and T cells in the TME is a known predictor of response to immunotherapy. In mouse models, tumours expressing the envelope protein of endogenous retroviruses show an upregulated immune response. This was noted at the RNA level, and retroviral proteins were also detected in the blood of patients with lung cancers, showing this phenomenon was mirrored in the clinic.
Commenting on the study, Downward, who was the senior author, said, “we now know that areas of B-cell expansion can help us predict a positive response to checkpoint inhibition, whereby we can boost B-cell activity in a targeted way for patients less likely to respond… this work opens up a number of new opportunities for improving patient responses to immunotherapy, a crucial step in helping more people survive lung cancer.”
Toxicity and resistance
The news is not all good, however. In lung cancer, it seems that patients who are sensitive to immunotherapies benefit from the addition of RAS inhibition. However, in those refractory to immunotherapy, the addition of RAS inhibitors does not rescue the response. Combining selective RAS inhibitors with immunotherapies also involves significant toxicity; considerable liver toxicity was reported, for instance, from a trial combining the anti-PD-1 pembrolizumab with sotorasib. Heavily mutated tumours – which RAS-driven non-small-cell lung cancers typically are – also have many escape mechanisms; resistance continues to be a key limiting factor in the success of these drug combinations.
It may therefore take some time before we have a clear idea of just how significant cracking the targeting of the G12C mutated KRAS turns out to be. Meanwhile, Shokat and others are busy exploring ways to target other common RAS mutations.
Learning the lessons
More than three decades of efforts have not yet solved the problem of curing or controlling RAS-driven cancers. However, a huge amount has been learned along the way about the complexity of cancer, and how the tumour microenvironment, coupled with clonal heterogeneity and subclonal resistance pathways, combine to thwart our best efforts to treat advanced disease.
Reflecting on his own perspective, Downward comments that, while the translational research landscape from basic biology to the clinic is much more dovetailed than when he first started in this field, the quality of that interaction still needs to be improved. Clinicians don’t always understand the importance of good clinical sampling, he feels, which impedes progress because the clinical samples used for research assessment are suboptimal.
He worries too that that the academic environment is less nurturing than when he started his career. Funding and publication pressures are impacting negatively on scientific progress. The scientific landscape has changed a lot, in ways that can often make a career in science seem less appealing. The volume and quantity of data that researchers need to keep up to date with to stay current is phenomenal, he adds, especially given the reproducibility crisis. And it is becoming almost impossible to repeat other’s work, in part because many experiments use company kits and methodologies that are not easy to troubleshoot if a problem occurs. Collectively, this adds up to quite a tough environment for newly qualified scientists, he says.
On a positive note, however, opportunities for working more collaboratively with experts in other fields have improved in the past 10 to 20 years, he notes, which gives younger scientists more options on how they carve out their career path. It also allows a greater exposure to other disciplines and fostering a broader skill set and knowledge base. Given what we now know about the inextricable links that tie the immune system to all cellular mechanisms that underpin cancer, that broad knowledge, skill set and collaboration will be essential to ensuring that the hard lessons of the past deliver successful treatments in the future.
Illustration by: Maddalena Carrai









