
Six lessons from the development of the first targeted anti-cancer therapy
Tamoxifen famously started life as a failed contraceptive, developed by ICI (now Astra Zeneca), but with the fatal flaw that it increased ovulation rather than suppressing it.
It’s a story sometimes told to show how discoveries can come from unexpected quarters and that scientific progress has a habit of proceeding in zigzags.
Craig Jordan is the pharmacologist who took that failed drug and developed a treatment strategy for tamoxifen, and then did the groundwork for four additional selective oestrogen receptor modulators (SERMs) – saving millions of women’s lives. He teaches us that such zigzaggedy progress and unexpected discoveries don’t just happen. They require bloody minded, dedicated and creative scientists who refuse to give up on failures, and fight for the resources and people to do the science to turn things that might not look very promising into something that could offer real value.
By ‘failures’, Jordan includes the vast majority of targeted medicines currently marketed with scant evidence of real benefit. “If you go to the paper that looks at the approvals of all of these targeted therapies, they have if you are lucky 5 or 10% responses. There is no survival data, but they are on the market,” (JAMA Onc 2018, 4:1093–98; JAMA Onc 2018, 4:1789–90).
Companies are able to sell them, says Jordan, on the basis that there’s nothing else, so why not use it? “My view is: why does nobody take the time and invest the money to find out which drugs will be used and useful?”
He believes that many failing targeted drugs would turn out to be of great value if more time and resources were invested in doing the pharmacology and translational research to build a detailed picture about exactly what they are doing and how they could be improved. That takes persistence (verging on obsession in his case) and a focus on finding solutions for patients rather than marketable applications for drugs.
You’re not going to cure cancer by sequencing everything
He worries that the major lesson to be learned from the tamoxifen/SERMs story won’t be learnt, partly because tamoxifen is seen by the younger generation of researchers as “a bit like aspirin” – part of the fabric of cancer therapy that has always been there. Moreover, while tamoxifen may be formally recognised as the first targeted anti-cancer therapy, it is Herceptin (trastuzumab) and Glivec (imatinib) that researchers generally use as a reference point, as they were the first to use new molecular biology techniques to identify targets and create molecules to block them.
For Jordan, this explosion of molecular biology is part of the problem. “Everybody could just go into the lab and take tumours and sequence them and compare them with the normal human genome. All we’ve really done is developed a map of the world. All of these maps. But nobody has a got a clue about: What is going on in Africa? Why is Detroit different from Los Angeles? What’s going on in Europe, and what are the interactions that create a European Union? Nobody has any idea because the task is so vast. You’re not going to cure cancer by sequencing everything.”
He says there has been a drift away from the fundamental questions: How does this drug work? How can we make it better? How do we study side effects? Could there be good side effects as well as bad? For Jordan, now professor of Breast Medical Oncology and Molecular and Cellular Oncology at MD Anderson Cancer Center, in Texas, the main drive was how to get a treatment available for women to keep them alive – and that focus seems to have got downgraded.
His one big exception to this critique relates to work on improving immunotherapies, including at MD Anderson, which is also home to Jim Allison, who received a Nobel Prize for his role in discovery of cancer therapy by inhibition of negative immune regulation. “There is a whole team of people looking at the good, bad and ugly of immunotherapy, and fixing it to be more targeted,” says Jordan. “Where do the side effects come from? Can we improve this? They are looking at everything that they can to be able to find out advances useful for patients. So this is a big version of the Craig Jordan model, if you like.”
Doing the pharmacology: lessons from tamoxifen
When Jordan started working with tamoxifen – then known as ICI 46 474 – in the 1970s, cancer researchers were betting heavily on the potential of combination chemotherapies to deliver a cure. They had had a dramatic effect on curing childhood leukaemia and great progress was being made in Hodgkin’s Disease. “Most of the clinical community were convinced you could find the right lexicon of drugs to give to any patient with any cancer and you could cure it.”

Breast cancer was a case in point, he says. “Heroic efforts were being made to try to treat women with massive doses of chemotherapy and bone marrow transplants, and none of this worked.” Tamoxifen, meanwhile, an anti-oestrogenic compound that had just failed as a contraceptive, “was just lying there”.
Jordan has described how, fresh from completing his PhD at the University of Leeds on the pharmacology of anti-oestrogens, he got the facilities (at the Worcester Foundation for Experimental Biology in the US) and the backing (from ICI in the UK) to explore the potential of tamoxifen in breast cancer – a story told in ‘Tamoxifen the first targeted long term adjuvant therapy for breast cancer’ (Endocr-Relat Cancer 2014, 21:R235–246) and ‘The SERM Saga, Something from Nothing’ (Ann Surg Oncol 2019, 26:1981–90).
However, it is what happened next, and over the following decades, that is probably more important for drawing lessons that can be applied to developing better cancer drugs today.
Lesson 1: How can this drug save lives? – a conversation with nature
The potential for treating breast cancer by cutting its access to oestrogen had been partly understood since the 1890s when George Beatson had shown that excising a woman’s ovaries could delay the progress of some breast cancers. However, ICI’s chief interest in developing ICI 46 474 was as a contraceptive, so until Jordan got the go-ahead to work on the drug in the early 1970s, not a single anti- tumour experiment had been done with it in the laboratory.
Arthur Walpole, who had been in charge of developing the agent as a contraceptive, but had a deep interest in cancer research, played a key role in convincing ICI not to ditch their drug at this point, but to advance it for approval as an orphan drug for use in advanced breast cancer. With his PhD background in anti-oestrogens, Jordan was tasked with doing the pharmacology to understand about its mechanism of action and clinical opportunities.
Tamoxifen’s impact in the metastatic setting, used across all tumours regardless of their hormonal status, was not spectacular. With a response rate of around 30% and a duration of response of around one to two years, it was no better than the hormonal therapies – high-dose oestrogen or androgen – that were already in use. The side effect profile was admittedly better, but the costs were higher, and ICI were not convinced there was money to be made with it.
But everything Jordan was learning about the drug was telling him that its true potential lay with a different strategy. His experiments with carcinogen-induced rat mammary cancers showed rapid tumour induction in controls, while those treated with tamoxifen remained completely tumour free: “Two depot injections of tamoxifen, which each had a biological action for many months, completely wiped out the development of tumours.”
He also showed that administering the tamoxifen sooner after inducing tumours with carcinogen was more effective than later, and that shorter duration delayed tumour development, but continuous administration had a long-term preventive effect (figure opposite).
The pharmacology, which Jordan describes as “a conversation with nature”, was telling him that the strength of tamoxifen lay in its potential as a preventive, or at least in very early disease. There were good reasons not to go down this route, however.
Firstly, prevention would mean giving the drug to healthy women who may never go on to develop the disease, so the side effects and risk would have to be negligible. Secondly, it is always more difficult to demonstrate efficacy in prevention than treatment, as prevention measures non-events. And anyway there was a high degree of scepticism within the research community about the efficacy of the drug, says Jordan.
The question was never: What is the easiest endpoint to prove? or What is the quickest route to market?
“They were saying ‘With chemotherapy women get sick, we see their blood cells go down, we know it’s killing cancer cells because it’s killing their healthy cells as well. This has virtually no side effects and you are saying use this because you think it will be able to kill cancer cells. You have no real evidence it will be able to do that.’”
For Jordan, however, the key question was never: What is the easiest endpoint to prove? Or what is the quickest route to a marketable drug? but rather: “What will keep women alive?”
Lesson 2: Face down the sceptics – nature does not lie
By the mid-1970s, and with Jordan now back in the UK, the concept of hitting cancer early was beginning to gain traction on both sides of the Atlantic, particularly in the form of adjuvant treatment following surgery for early breast cancer. “My philosophy was that it is no good trying to cure people at the end of life. You’ve got to hit it strategically somewhere along the way that will become vulnerable. That is after a woman had had a mastectomy and there are micrometastases around her body but we can’t see them.”
Nature says… continuous tamoxifen is the way to go
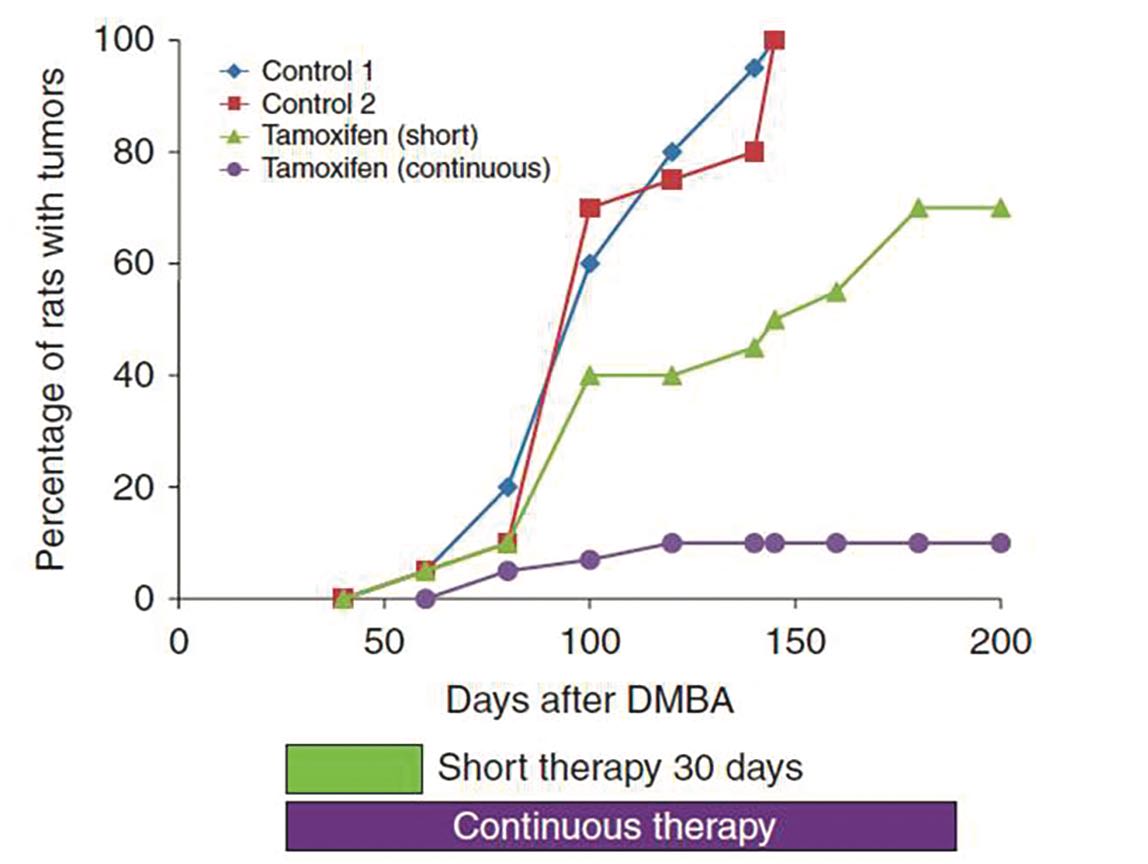
Source: VC Jordan (2014) Endocrine-Related Cancer 21:R235–R246, republished with permission
In September 1977, at a packed Breast Cancer Symposium at King’s College Cambridge, Jordan presented his data showing that, in rats, a continuous dose of tamoxifen could offer long-term protection against breast cancer. It was here that he first argued the case for the potential of long-term use of tamoxifen in an adjuvant setting. The suggestion did not go down well.
They were horrified, says Jordan, and protested that he wasn’t a doctor, didn’t understand anything about drug resistance, and presented a danger. “It was completely counterintuitive in cancer, having a therapy you give for ever. Everyone said it can’t happen.”

Everything that clinicians had learnt about the limitations of systemic therapies – including tamoxifen, which in advanced cancers stopped working after a year or two – pointed to the intractable problem of acquired resistance, says Jordan. The dominant feeling in the audience – with a few important exceptions – was that a strategy that risked developing resistance in an adjuvant setting meant having one less option to use if the woman then went on to develop advanced disease.
But Jordan’s pharmacology was telling him long-term adjuvant treatment was the way to go. Undeterred, he took his data to the US, where he presented his case to Paul Carbone, who was setting up the Wisconsin Comprehensive Cancer Center, one of six original comprehensive cancer centres designated by the National Cancer Institute.
Here he met a very different response. Carbone invited him to join them, and head the breast cancer programme.
Lesson 3: Find the right research environment
The labs at Wisconsin became the cradle where the concept of selective oestrogen receptor modulators was developed, where raloxifene, the second SERM after tamoxifen was developed, and where early work led to three further SERMs, each addressing multiple key women’s health issues.
The impact of tamoxifen in advanced breast cancers gave no clue about its amazing value in an adjuvant setting
Here, for the first time Jordan was based at an institute that treated patients, where clinicians and pharmacologists learned together via feedback loops between lab and clinic – long before the term ‘translational research’ was coined. The very significant public funding made available as part of the ‘War on Cancer’ gave researchers the freedom to pursue scientific strategies led by seeking solutions for patients, without constant pressure to demonstrate marketable applications for a drug, take out patents, or spin off biotech companies.
So began the interaction with clinical trials organisations such as the National Surgical Adjuvant Breast and Bowel Project (NSABP) led by Bernie Fisher, a key instigator of the trials of adjuvant chemotherapy, but also with triallists in the UK, such as Michael Baum – who was the first to trial adjuvant tamoxifen for two years rather than one (the strategically named NATO trial) – and Helen Stewart, who ran the Scottish Adjuvant Tamoxifen trial that looked at the benefit of administering the drug for five years immediately after mastectomy compared with waiting until recurrence.
The results of the Scottish trial showed “significantly prolonged disease-free survival” in the adjuvant arm for patient population.
It was not until more than 10 years later, however, with publication of the 1998 Oxford Overview Analysis – a meta-analysis of data from multiple trials done on adjuvant breast cancer therapy at that time – that the true size of that benefit became clear. In premenopausal women whose tumours were oestrogen receptor positive, adjuvant tamoxifen given for one year produced no reduction in recurrence or death rates. Two years’ administration produced a small benefit on both measures. Five years gave an astonishing 50% reduction in recurrence and a 30% reduction in death rates.
If there’s one lesson that Jordan wants to get across, it is this. The impact of tamoxifen used in advanced breast cancers gave no clue about its amazing potential given long-term in an adjuvant setting. “Nobody could have predicted that at all.”
Winning the argument on the issue of resistance was an important key to that success – we now know that tamoxifen can continue to be administered for 10 years or more. Demonstrating that tamoxifen was a precision treatment that should only be used – and its value measured – in women with hormone-dependent breast cancer, was also key.
Lesson 4: Mechanism of action – how tamoxifen became the first precision cancer therapy
Jordan understood from the start that the oestrogen receptor was likely to play a key role in tamoxifen’s mechanism of action. In 1973 Elwood Jensen, who had identified the receptor some 17 years earlier, offered him the chance to visit Chicago to learn assay techniques. By 1975 Jordan’s labs were able to show that tamoxifen blocks estradiol binding to human tumour oestrogen receptors.
The pharmacology told him that, unlike cytotoxics, which target every replicating cell, tamoxifen would work only in certain breast cancers – those with high levels of oestrogen receptors. This was one of the first indications that that biological drivers behind breast cancer might not all be the same. But it took another 10 years before the concept of testing and selective administration was widely accepted, says Jordan.
One reason for the delayed recognition was that the pharmacological findings had not been confirmed in the original Scottish trial, or the NATO trial, probably due to a lack of preparation of the tissue before it got to the lab, which destroyed the oestrogen receptor.
By the early 1990s, though, testing breast tumours for their hormonal status became routine, signalling the arrival of the concept of precision cancer medicine.

Lesson 5: Search for ‘the good, the bad and the ugly’
Tamoxifen was now a block buster drug delivering unparalleled benefit to hundreds of thousands of women across the world. At this point, Jordan decided to do something that drug sponsors never do: he and his lab went back to the molecule to find out everything they could.
“I was trained as a pharmacologist to look for the good, the bad and the ugly. You’ve got to be able to spot what is going to go wrong, so people do not die. We took it apart like nobody else had taken it apart. Nobody else was interested. The drug was on the market. Who cared?”
He had won the argument about long-term administration of a cancer drug. He would now take responsibility for exploring every aspect of its impact, to look for potential side effects and drivers of resistance.
That is how Jordan – having devoted his career to a drug he believed in and steered to success – ended up as the person who sounded the alert over the raised risk of endometrial cancer associated with taking tamoxifen.
Jordan’s lab had noticed that the drug had a uterine ‘tickle’, inducing small changes, principally a thickening of the uterine wall. To find out more, his lab conducted experiments with immune-deficient mice, implanting human cancer cell lines, injecting oestrogen to make them grow, and then administering tamoxifen. This time they introduced a breast cancer cell line on one side of the mouse, and an endometrial cancer cell line on the other. “Here was the revelation. The breast cancer was completely blocked by tamoxifen. Gone. But the animal was dragging around a huge endometrial cancer.”
SERMs concept and consequences in cancer
Nothing was being flagged up in the clinical setting, however, so Jordan found it hard to get his concerns taken seriously. Taking matters into his own hands, he decided to announce his findings at an international meeting he was due to address. His findings prompted a doctor, who was sitting in the audience, to report some clinical cases of endometrial cancers in women he had treated with tamoxifen.
A correspondence then opened in the pages of The Lancet between the doctor, Leonard Hardell, and Jordan. Jordan suggested checking the Scottish trial data for raised incidence of endometrial cancers. “Never seen it!” was the response, says Jordan. But then the Scottish trial, reported in The Lancet in 1987, had never actually collected data on the incidence of endometrial cancers. In the end it was a Scandinavian clinical trials group cancer registry that delivered the smoking gun. “They looked at their data gathered on five years of tamoxifen versus two years versus control, and said we were right… They had nine years of data, but they hadn’t looked at it.”
Yet again, says Jordan, it was the pharmacological work and not the clinical data that brought this essential information to light. Subsequent studies revealed that the raised endometrial risk only affected women post-menopause, as monthly menstruation is a protective factor.
This finding destroyed Jordan’s hopes of a chemopreventive role for tamoxifen in postmenopausal women. But it did mean that, by the time chemoprevention trials on tamoxifen derivatives began, researchers were looking at the whole gynaecological picture. This was due to the work of his lab. “I consider it one of the best things I have ever done,” he says.
Lesson 6: Can we do better? Raloxifene and more…
Painstaking exploration of ‘the good, the bad and the ugly’ had revealed not only the heightened risk of endometrial cancers, it also revealed some potentially ‘good’ and completely counterintuitive effects of tamoxifen.
Tamoxifen was only selectively anti-oestrogenic, and actually acted as an oestrogen agonist in some instances. “It would switch on and switch off sites around the woman’s body that nobody had ever seen before,” says Jordan. “Anti-oestrogen was thought to be anti-oestrogenic everywhere in the body, so it would cause osteoporosis, it would cause coronary heart disease… What we found was that tamoxifen tickles the bones to make them stay strong, and it lowered circulating cholesterol.”
He developed the idea of looking for derivatives of tamoxifen that might carry no risk of endometrial cancer, but could treat coronary heart disease (a bigger killer than breast cancer among women) and treat osteoporosis (another major killer of older women, due to complications of fractures), while preventing breast cancer at the same time.
That is how the concept of selective oestrogen receptor modulators, or SERMs, was born in 1989, with the ability to “treat multiple diseases with one pill”.
Being the ‘go-to’ lab for all things related to oestrogen receptors and anti-oestrogen, over the years Jordan had accumulated a number of tamoxifen-like synthetic compounds that he’d been asked to test against tamoxifen. He now turned his attention to pulling together information about this group of drugs.
Persistence and the deep knowledge accumulated over 30 years specialising in this area, paid off in 1997 with approval of raloxifene – the second SERM discovered in Jordan’s lab – for treating osteoporosis. The drug carried none of the risk of endometrial cancer associated with tamoxifen but gave protection (though only 75% as effectively as tamoxifen) against breast cancer when taken continuously.
The start of SERM chemistry
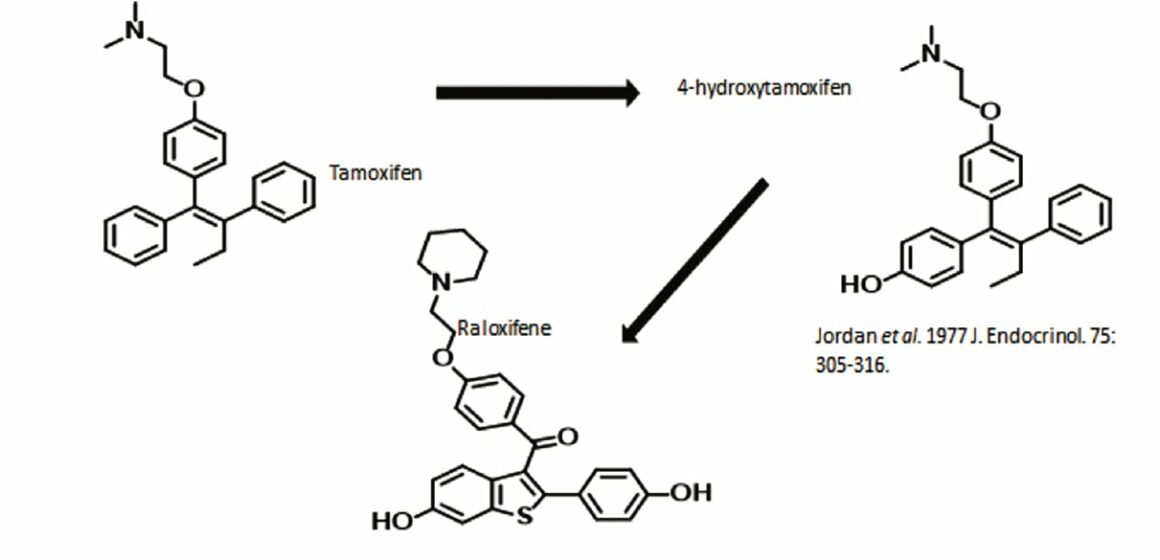
Source: VC Jordan et al (1977) J Endocrinol 75:305–16
A theroretical study, ten years later, that compared rates of new breast cancers among women treated for osteoporosis with raloxifene compared to the then-standard hormone replacement therapy (HRT), or bisphosphonates, showed how effective this strategy was. Applying the different rates to the 500,000 women estimated to have been treated with raloxifene across the world, analysis suggested that over a ten-year period, about 27,000 breast cancers were being prevented as a side effect of active treatment for osteoporosis – a marked success for the chemoprevention strategy Jordan had always believed in (EJC 2006, 42:2909–13)
In 2007, the FDA extended the indication for raloxifene for primary use in preventing breast cancer for women known to be at particularly high risk.
Three further related SERMs have since come to market: bazedoxifene – approved in Europe and the US as part of a treatment for vasomotor symptoms associated with menopause and the prevention of postmenopausal osteoporosis; ospemifene – approved on both sides of the Atlantic for the treatment of symptoms of vulvar and vaginal atrophy due to menopause; and lasofoxifene. The last of these – “an old drug from the contraceptive days,” and “a miracle of medicinal chemistry”, according to Jordan – decreases fractures from osteoporosis using 1% of the dose required for equivalent impact with raloxifene, while also reducing breast cancer, stroke and – a first for any SERM – coronary heart disease, though with increased risk for venous thromboembolic events.
Sadly, says Jordan, due to the intricacies of pharma marketing strategies, lasofoxifene has been left sitting on the shelf, approved but not marketed in Europe, and not even approved yet in the US.
Can drug development get back on track?
A 2014 review on Past, Present and Future Challenges in Breast Cancer Treatment, written by a star cast of authors and published to mark the 50th anniversary of ASCO (the American Society of Clinical Oncology), claimed that anti-oestrogen treatments had arguably had “greater global impact that any other treatment intervention in cancer medicine,” (JCO 2014, 32:1979–86).

Photo: PA © PA
The reference included not just tamoxifen and its derivatives, which work through selective modulation of oestrogen receptors, but also aromatase inhibitors, a newer class of drugs introduced in the mid-2000s that work by suppressing oestrogen production.
The value of tamoxifen itself, says Jordan, can be measured by the fact that 25 years on it has not been replaced, and is still used as treatment for advanced disease, as an adjuvant in early disease, as a treatment for ductal carcinoma in situ, as chemoprevention in high-risk premenopausal women, and in male breast cancer. “No other therapy has that penetration in cancer across the board.”
Contrast this with the drugs that have come on the market in the age of molecular biology and precision cancer medicine. A 2017 study in the British Medical Journal reported that, from 2009 to 2013, the European Medicines Agency approved 48 cancer drugs for 68 indications. Of the 44 drug indications that did not show a survival benefit at time of approval, and with a median of 5.4 years’ follow up (3.3–8.1 yrs), three (7%) were subsequently shown to extend life after market entry, and five (11%) were associated with some improvements in quality of life (BMJ 2017, 359:j4530).
Jordan argues that one big factor is that governments have ceded the task of drug development to the private sector, “which is expected to raise private capital and get it done”. The public money that funded much of his early work at the University of Leeds and at Wisconsin has all but dried up. In the US, he says, only around 1 in 15 young scientists can get a grant today – in his day it was 1 in 4. “[Governments argue] Why should we fund the research? Go out and start your own biotech company and raise private capital and get it done.”
Pharmaceutical companies have also stopped doing their own drug development work. “Places like [ICI/Astra Zeneca’s] Alderley Park, which developed dozens and dozens of world beating drugs, all that has been closed down.” Biotechs, meanwhile, measure their success in terms of coming up with “an idea that looks like it has promise – that big pharma will buy.”
But at no point is anyone investing in the pharmacological work to turn that promise into a really effective drug, says Jordan.
Although targeted therapies go to a specific gene target, he says, there are no tests to put the gene target and response to the therapy together. “Nobody is doing that. It is ‘suck it and see’ with every one of them. We’ve gone back to the days before the oestrogen receptor and SERMs.”
He sees Glivec and Herceptin as rare exceptions “[They are] the benchmarks of the past generation. But now we have 200 different targeted drugs and where do you start?
“Now I think it has all gone so far adrift, into what we can find from the sequencing machine, that we have lost the skills to be able to ask the questions about how is this treatment going to impact on this disease, and what is the good, bad and ugly of my new drug?”









